ID intern unic: 353339
Версия на русском

Republica Moldova
din 04.06.2014
pentru aprobarea Regulamentului privind condiţiile de
plasare pe piaţă a dispozitivelor medicale implantabile active
plasare pe piaţă a dispozitivelor medicale implantabile active
Abrogată prin HG704 din 11.07.18, MO336-346/07.09.18 art.902
În temeiul prevederilor art. 54 din Legea ocrotirii sănătăţii nr. 411-XIII din 28 martie 1995 (Monitorul Oficial al Republicii Moldova, 1995, nr. 34, art. 373), cu modificările şi completările ulterioare, art. 4 alin. (4), art. 5, art. 13, art. 21 alin. (3), art. 25 alin. (1) din Legea nr. 92 din 26 aprilie 2012 cu privire la dispozitivele medicale (Monitorul Oficial al Republicii Moldova, 2012, nr. 149-154, art. 480), Guvernul HOTĂRĂŞTE:
1. Se aprobă Regulamentul privind condiţiile de plasare pe piaţă a dispozitivelor medicale implantabile active (se anexează).
2. Controlul asupra executării prezentei hotărîri se pune în sarcina Ministerului Sănătăţii şi Ministerului Mediului.
PRIM-MINISTRU Iurie LEANCĂ
Contrasemnează:
Viceprim-ministru,
ministrul economiei Valeriu Lazăr
Ministrul sănătăţii Andrei Usatîi
Ministrul mediului Gheorghe Şalaru
Nr. 410. Chişinău, 4 iunie 2014.
2. În sensul prezentului Regulament se utilizează terminologia definită în Legea nr. 92 din 26 aprilie 2012 cu privire la dispozitivele medicale, Legea nr. 235 din 1 decembrie 2011 privind activităţile de acreditare şi de evaluare a conformităţii, Legea nr. 1409-XIII din 17 decembrie 1997 cu privire la medicamente, precum şi următoarea noţiune:
organism de evaluare a conformităţii recunoscut – organism de evaluare a conformităţii acreditat, persoană juridică cu sediul în Republica Moldova şi recunoscut de Ministerul Sănătăţii pentru activitatea de evaluare a conformităţii, conform actelor normative cu privire la dispozitivele medicale, aprobate de Guvern.
3. În cazul în care un dispozitiv medical implantabil activ este destinat administrării unui medicament în sensul definiţiei din art. 2 din Legea nr. 92 din 26 aprilie 2012 cu privire la dispozitivele medicale, acesta este reglementat prin prezentul Regulament, fără a aduce atingere definiţiei privitor la medicamente.
4. În cazul în care un dispozitiv medical implantabil activ încorporează, ca parte integrantă, o substanţă care, dacă este utilizată separat, se consideră medicament în sensul art. 3 din Legea nr. 1409-XIII din 17 decembrie 1997 cu privire la medicamente şi care acţionează asupra organismului uman printr-o acţiune auxiliară celei a dispozitivului, acest dispozitiv se evaluează şi se înregistrează conform prevederilor prezentului Regulament.
5. În cazul în care un dispozitiv medical implantabil activ încorporează, ca parte integrantă, o substanţă care, dacă este utilizată separat, se consideră constituent al unui medicament sau un medicament derivat din sînge uman sau din plasmă umană (în continuare – derivat din sînge uman) şi care poate acţiona asupra organismului uman printr-o acţiune auxiliară celei a dispozitivului, acesta este evaluat şi înregistrat în conformitate cu prevederile prezentului Regulament.
6. Prezentul Regulament constituie o reglementare specifică în sensul pct.4 din Reglementarea tehnică „Compatibilitatea electromagnetică a echipamentelor”, aprobată prin Hotărîrea Guvernului nr. 95 din 4 februarie 2008 „Cu privire la aprobarea Reglementării tehnice „Compatibilitatea electromagnetică a echipamentelor”.
7. Prezentul Regulament nu se aplică:
1) medicamentelor;
2) sîngelui uman, produselor din sînge, plasmei sau celulelor sangvine de origine umană ori dispozitivelor care încorporează în momentul introducerii lor pe piaţă astfel de produse din sînge, plasmă sau celule, cu excepţia dispozitivelor prevăzute la pct. 5 din prezentul Regulament;
3) transplanturilor, ţesuturilor sau celulelor de origine umană, precum şi produselor care încorporează sau derivă din ţesuturi ori celule de origine umană, cu excepţia dispozitivelor prevăzute la pct. 5 din prezentul Regulament;
4) transplanturilor, ţesuturilor sau celulelor de origine animală, cu excepţia cazurilor în care un dispozitiv este fabricat prin utilizarea de ţesuturi de origine animală neviabile sau de produse neviabile derivate din ţesuturi de origine animală.
9. Dispozitivele medicale implantabile active (în continuare – dispozitive) la care se face referire în pct. 2-5 din prezentul Regulament asigură conformitatea cu cerinţele esenţiale prevăzute în anexa nr. 1 la prezentul Regulament, care le sînt aplicabile, conform scopului propus al acestora.
10. Se introduc pe piaţă şi se pun în funcţiune numai dispozitivele conforme cu prevederile prezentului Regulament şi care poartă marcajul european de conformitate CE, prevăzut la pct. 42-44 din prezentul Regulament, care demonstrează că aceste dispozitive au fost supuse evaluării conformităţii potrivit capitolului V din prezentul Regulament.
11. Nu pot face obiectul restricţionării:
1) punerea la dispoziţia medicilor calificaţi în mod corespunzător sau a persoanelor autorizate în acest scop a dispozitivelor destinate investigaţiilor clinice, dacă acestea îndeplinesc condiţiile prevăzute în capitolul VI şi în anexa nr. 6 la prezentul Regulament;
2) introducerea pe piaţă şi punerea în funcţiune a dispozitivelor fabricate la comandă, dacă acestea îndeplinesc condiţiile prevăzute în anexa nr. 6 la prezentul Regulament şi sînt însoţite de declaraţia menţionată în această anexă, care se pune la dispoziţia pacientului specific identificat. Dispozitivele menţionate nu poartă marcajul CE de conformitate.
12. La tîrgurile, expoziţiile, demonstraţiile, întrunirile ştiinţifice şi tehnice şi altele asemenea organizate pe teritoriul Republicii Moldova, dispozitivele care nu sînt conforme cu prevederile prezentului Regulament pot fi expuse, cu condiţia să poarte o inscripţionare vizibilă, care să indice în mod clar că nu pot fi comercializate sau puse în funcţiune înainte de a fi aduse la conformitate cu prevederile prezentului Regulament.
13. Dacă un dispozitiv este pus în funcţiune, informaţiile prevăzute la pct. 11 alin. 8) şi 9) şi pct. 12 din anexa nr. 1 la prezentul Regulament urmează să fie furnizate în limba de stat.
14. În cazul în care un dispozitiv face obiectul mai multor reglementări tehnice care prevăd aplicarea marcajului de conformitate, marcajul semnifică faptul că dispozitivul este conform cu prevederile tuturor reglementărilor tehnice respective.
15. Dacă una sau mai multe dintre reglementările tehnice prevăzute la pct. 12 permit producătorului, pentru o perioadă tranzitorie, să aleagă reglementările pe care să le aplice, marcajul de conformitate semnifică faptul că dispozitivele satisfac numai prevederile reglementărilor aplicate de producător.
16. În cazul prevăzut la pct.13 din prezentul Regulament, elementele de identificare ale reglementărilor tehnice aplicate de producător se indică în documentele, notele sau instrucţiunile cerute de reglementările care însoţesc dispozitivul. Documentele, notele sau instrucţiunile care însoţesc dispozitivul trebuie să fie accesibile, fără a fi necesară distrugerea ambalajului care asigură sterilitatea dispozitivului.
17. Se consideră, că cerinţele esenţiale prevăzute în anexa nr. 1 la prezentul Regulament sînt îndeplinite, dacă dispozitivele medicale sînt conforme cu specificaţiile tehnice din standardele naţionale conexe la prezentul Regulament, care adoptă standardele europene armonizate.
În sensul prezentului Regulament, referirea la standardele armonizate include, de asemenea, monografiile Farmacopeii Europene, în special în ceea ce priveşte interacţiunea dintre medicamente şi materialele utilizate în dispozitivele care conţin astfel de medicamente, ale căror referinţe au fost publicate în Monitorul Oficial al Republicii Moldova.
În situaţia în care Agenţia constată că standardele naţionale conexe nu satisfac în totalitate cerinţele esenţiale prevăzute în anexa nr. 1 la prezentul Regulament, aceasta va aplica una din prevederile art. 62 din Legea nr. 590-XIII din 22 septembrie 1995 cu privire la standardizare.
Producătorul este responsabil pentru activităţile ulterioare retragerii/interzicerii dispozitivelor medicale.
19. Agenţia informează producătorul sau reprezentantul autorizat al acestuia, importatorul/distribuitorul, instituţiile medico-sanitare sau alţi utilizatori cu privire la măsurile prevăzute la pct. 18 din prezentul Regulament, indicînd motivele pentru care a luat această decizie şi, în special, dacă neconformitatea cu prezentul Regulament se datorează următoarelor cauze:
1) neîndeplinirea cerinţelor esenţiale menţionate în anexa nr. 1 la prezentul Regulament, în cazul în care dispozitivul nu îndeplineşte în totalitate sau în parte standardele prevăzute la pct. 17 alineatul unu din prezentul Regulament;
2) aplicarea incorectă a standardelor menţionate la pct. 17 alineatul unu din prezentul Regulament;
3) unele deficienţe ale standardelor.
20. În cazul în care dispozitivele medicale necorespunzătoare poartă marcajul CE, Agenţia are obligaţia de a anunţa Comisia Europeană în termen de 72 de ore de la data constatării.
1) orice funcţionare defectuoasă sau deteriorare a caracteristicilor şi a performanţelor unui dispozitiv, precum şi orice etichetare sau instrucţiuni de utilizare inadecvate, care pot să conducă ori au condus la decesul sau deteriorarea severă a stării de sănătate a unui pacient ori utilizator;
2) orice cauză de ordin tehnic sau medical vizînd caracteristicile ori performanţele unui dispozitiv, care, din motivele prevăzute la subpct. 1) din prezentul punct, conduce la retragerea sistematică de pe piaţă de către producător a dispozitivelor de acelaşi tip.
22. Obligaţia de a anunţa Agenţia despre incidentele menţionate la pct. 21 revine producătorului sau reprezentantului său autorizat, importatorului, distribuitorului, personalului medical, instituţiilor medico-sanitare sau altor utilizatori.
23. În cazul în care informarea despre incidentele menţionate la pct. 21 a fost transmisă de personalul medical, instituţiile medico-sanitare sau de către alţi utilizatori, Agenţia informează producătorul dispozitivului în cauză ori reprezentantul său autorizat cu privire la incident.
24. După efectuarea unei evaluări, dacă este posibil, împreună cu producătorul sau cu reprezentantul său autorizat, respectînd prevederile capitolului III din prezentul Regulament, Agenţia informează imediat autorităţile competente din alte state cu care are încheiate acorduri referitor la măsurile care au fost luate sau care sînt avute în vedere pentru minimizarea riscului de reproducere a incidentelor menţionate la pct. 21, inclusiv informaţii referitoare la incidentele depistate.
1) procedura referitoare la declaraţia de conformitate, prevăzută în anexa nr. 2 la prezentul Regulament;
2) procedura referitoare la examinarea CE de tip, prevăzută în anexa nr. 3 la prezentul Regulament, asociată cu:
a) procedura referitoare la verificarea unităţii de produs, prevăzută în anexa nr. 4 la prezentul Regulament;
b) procedura referitoare la declaraţia de conformitate, prevăzută în anexa nr. 5 la prezentul Regulament.
26. În cazul dispozitivelor fabricate la comandă, producătorul trebuie să emită declaraţia privind dispozitivele cu scopuri speciale, conform anexei nr. 6 la prezentul Regulament, înainte de introducerea pe piaţă a fiecărui dispozitiv.
27. În cazuri argumentate, procedurile prevăzute în anexele nr. 3, 4 şi 6 la prezentul Regulament se aplică de reprezentantul autorizat al producătorului stabilit în Republica Moldova.
28. Înregistrările şi corespondenţa referitoare la procedurile menţionate la pct. 25-27 din prezentul Regulament se redactează în limba de stat.
29. Evaluarea conformităţii dispozitivelor medicale se efectuează de organisme de evaluare a conformităţii, acreditate în condiţiile Legii nr. 235 din 1 decembrie 2011 privind activităţile de acreditare şi de evaluare a conformităţii şi recunoscute de Ministerul Sănătăţii, în baza criteriilor stabilite de prezentul Regulament.
În cursul procedurii de evaluare a conformităţii pentru un dispozitiv, producătorul şi/sau organismul de evaluare a conformităţii recunoscut (în continuare – organism recunoscut) ţine cont de rezultatele obţinute în urma oricăror operaţiuni de evaluare şi verificare care au fost efectuate potrivit prevederilor prezentului Regulament într-o fază intermediară de fabricaţie.
30. În cazul în care procedura de evaluare a conformităţii implică intervenţia unui organism recunoscut, producătorul sau reprezentantul său autorizat stabilit în Republica Moldova este în drept să se adreseze unui organism la alegere, corespunzător sarcinilor în legătură cu care acesta a fost recunoscut.
31. Organismul recunoscut solicită orice informaţii sau date suplimentare care sînt necesare pentru a stabili şi a menţine atestarea conformităţii în funcţie de procedura aleasă.
32. Deciziile adoptate de organismul recunoscut potrivit prevederilor anexelor nr. 2, 3 şi 5 la prezentul Regulament au o valabilitate maximă de 5 ani şi urmează a fi prelungite, pentru perioade suplimentare de cel mult 5 ani, la cererea înaintată de către producătorul sau reprezentantul său autorizat, la o dată stabilită în contractul semnat de ambele părţi.
33. Agenţia autorizează, în cazuri deosebite (cataclisme, catastrofe, epidemii, epizootii, intoxicaţii în masă, în alte cazuri ce ameninţă sănătatea oamenilor; absenţa pe piaţă a analogurilor sau a substituenţilor dispozitivelor medicale), punerea la dispoziţie pe piaţă şi punerea în funcţiune a dispozitivelor neautorizate în Republica Moldova, dar autorizate în ţara de origine, pentru care nu au fost efectuate procedurile prevăzute la pct. 27-31 din prezentul Regulament şi a căror utilizare este în interesul protecţiei sănătăţii.
După expirarea perioadei de forţă majoră, dar nu mai mult de 1 an de la declanşarea acesteia, dispozitivele sus-menţionate urmează să fie supuse în mod obligatoriu procedurii de autorizare în Republica Moldova. În caz contrar, Agenţia îşi rezervă dreptul de a dispune retragerea de pe piaţă şi de a interzice plasarea pe piaţă a dispozitivelor respective.
34. Agenţia asigură realizarea acţiunilor în următoarele situaţii:
1) atunci cînd constată că stabilirea conformităţii unui dispozitiv sau a unei grupe de dispozitive se efectuează prin excepţie de la prevederile capitolului V din prezentul Regulament, prin aplicarea exclusivă a unei proceduri respective, selectată dintre cele prevăzute la capitolul V din prezentul Regulament;
2) atunci cînd constată că este necesară o decizie pentru a determina dacă un anumit produs sau grup de produse se încadrează în definiţia prevăzută în Legea nr. 92 din 26 aprilie 2012 cu privire la dispozitivele medicale: „dispozitiv medical”, „dispozitiv medical implantabil activ”, „dispozitiv fabricat la comandă” şi „dispozitiv destinat investigaţiei clinice”.
36. Producătorul este în drept să înceapă investigaţiile clinice la sfîrşitul perioadei de 60 de zile de la notificare, doar dacă pînă la sfîrşitul acestei perioade Agenţia îi comunică acestuia decizia de acceptare.
Agenţia va autoriza producătorii pentru începerea investigaţiilor clinice respective înaintea expirării perioadei de 60 de zile, dacă Comisia pentru dispozitive medicale a emis un aviz favorabil privind programul investigaţiei în cauză, cuprinzînd şi analiza sa referitoare la planul investigaţiei clinice.
37. Agenţia are obligaţia să asigure sănătatea publică şi realizarea reglementărilor în domeniu:
1) în cazul în care investigaţia clinică este refuzată sau suspendată de Agenţie, ultima comunică solicitantului decizia sa şi motivele care au stat la baza acesteia;
2) în cazul în care Agenţia a solicitat o modificare semnificativă sau întreruperea temporară a unei investigaţii clinice, aceasta informează solicitantul cu privire la acţiunile sale şi motivele pentru acţiunile întreprinse.
38. Producătorul sau reprezentantul său autorizat notifică Agenţia cu privire la sfîrşitul investigaţiei clinice, cu o justificare în cazul unei încetări anticipate.
În cazul unei încetări anticipate a investigaţiei clinice din motive de securitate, Agenţia comunică solicitantului această notificare.
Producătorul sau reprezentantul său autorizat pune la dispoziţia Agenţiei raportul prevăzut în secţiunea a II-a, pct.3, subpct. 7) din anexa nr. 7 la prezentul Regulament.
39. Investigaţiile clinice se desfăşoară potrivit prevederilor anexei nr. 7 la prezentul Regulament.
41. Pentru toate dispozitivele, Agenţia se informează cu privire la toate datele care permit identificarea acestor dispozitive, împreună cu eticheta şi instrucţiunile de utilizare, atunci cînd dispozitivele sînt puse în funcţiune pe teritoriul Republicii Moldova.
42. În cazul în care un producător care introduce pe piaţă un dispozitiv în nume propriu nu are sediu juridic în Republica Moldova, acesta desemnează un reprezentant autorizat în Republica Moldova.
43. În cazul dispozitivelor prevăzute la pct. 40 din prezentul Regulament, reprezentantul autorizat cu sediu juridic în Republica Moldova informează Agenţia cu privire la toate datele menţionate la pct. 40 din prezentul Regulament.
44. Agenţia informează, la cerere, autorităţile competente din alte state cu care are încheiate acorduri cu privire la datele prevăzute la pct. 40 din prezentul Regulament, furnizate de către producător sau de către reprezentantul său autorizat.
45. Datele înregistrate, potrivit prevederilor prezentului Regulament, se stochează în baza de date privind dispozitivele medicale a Agenţiei.
46. Baza de date privind dispozitivele medicale a Agenţiei cuprinde informaţii referitoare la:
1) înregistrarea producătorilor şi dispozitivelor, potrivit prevederilor pct. 40;
2) certificatele emise, modificate, suplimentate, suspendate, retrase sau respinse potrivit prevederilor din anexele nr. 2-5 la prezentul Regulament;
3) procedura de vigilenţă prevăzută în capitolul IV la prezentul Regulament;
4) investigaţiile clinice prevăzute în capitolul VI din prezentul Regulament.
Datele prevăzute la prezentul punct se furnizează în format standard.
47. Normele de procedură pentru aplicarea prevederilor pct. 45-46 din prezentul Regulament se aprobă prin ordin al Ministerului Sănătăţii.
49. Agenţia informează despre măsurile aplicate potrivit pct. 48 Ministerul Sănătăţii, producătorul şi organismul de evaluare a conformităţii recunoscut.
În cazul în care Agenţia constată că un organism recunoscut nu corespunde criteriilor specificate, care au stat la baza recunoaşterii, propune Ministerului Sănătăţii anularea ordinului de recunoaştere şi informează Centrul Naţional de Acreditare.
51. Organismul recunoscut şi producătorul sau reprezentantul său autorizat stabilesc de comun acord termenele-limită pentru finalizarea activităţilor de evaluare şi verificare prevăzute în anexele nr. 2-5 la prezentul Regulament.
52. Organismul recunoscut informează Agenţia referitor la toate certificatele emise, modificate, suplimentate, suspendate, retrase sau refuzate, precum şi celelalte organisme recunoscute în sensul prezentului Regulament cu privire la certificatele retrase, suspendate sau refuzate şi, la cerere, cu privire la certificatele eliberate. Organismul recunoscut oferă, de asemenea, la cererea Agenţiei, toate informaţiile suplimentare relevante.
53. În cazul în care un organism recunoscut constată că cerinţele relevante din prezentul Regulament nu au fost îndeplinite sau au încetat să mai fie îndeplinite de producător sau că un certificat nu ar fi trebuit să fie emis, atunci, bazîndu-se pe principiul proporţionalităţii, suspendă sau retrage certificatul emis ori impune restricţii asupra acestuia pînă cînd conformitatea cu aceste cerinţe este asigurată de către producător.
În cazul suspendării sau retragerii certificatului ori al impunerii de restricţii sau în cazurile în care este necesară o intervenţie din partea autorităţii competente, organismul recunoscut informează Agenţia cu privire la acest fapt.
Agenţia informează părţile interesate cu privire la măsurile luate, conform alineatului unu din prezentul punct.
54. Organismul recunoscut oferă, la cerere, orice informaţii sau documente relevante, inclusiv documentele bugetare, necesare pentru a-i permite Agenţiei să verifice respectarea criteriilor menţionate în anexa nr. 8 la prezentul Regulament.
56. Marcajul de conformitate, potrivit prevederilor anexei nr. 9 la prezentul Regulament, se aplică vizibil, lizibil şi de neşters pe ambalajul steril şi, dacă este cazul, pe ambalajul comercial şi pe instrucţiunile de utilizare.
Marcajul de conformitate este însoţit de numărul de identificare al organismului recunoscut, care poartă răspunderea pentru aplicarea procedurilor prevăzute în anexele nr. 2, 4 şi 5 la prezentul Regulament.
57. Este interzisă aplicarea de marcaje care poate induce în eroare părţile terţe cu privire la înţelesul sau forma grafică a marcajului de conformitate.
Se aplică, de asemenea, orice alt marcaj pe ambalaj sau pe instrucţiunile care însoţesc dispozitivul, cu condiţia ca acesta să nu afecteze vizibilitatea şi claritatea marcajului de conformitate.
58. Fără a aduce atingere prevederilor capitolului III din prezentul Regulament, în cazul în care Agenţia stabileşte că marcajul de conformitate a fost aplicat în mod necorespunzător sau lipseşte, încălcînd prezentul Regulament, producătorul sau reprezentantul său autorizat este obligat să lichideze încălcările depistate.
Dacă se menţine situaţia de neconformitate prevăzută la alineatul unu din prezentul punct, Agenţia limitează sau interzice introducerea pe piaţă a produsului în cauză ori se asigură că este retras de pe piaţă, în conformitate cu procedura prevăzută la capitolul III din prezentul Regulament.
59. Prevederile pct. 58 din prezentul Regulament se aplică şi atunci cînd marcajul de conformitate a fost aplicat produselor care nu fac obiectul prezentului Regulament, dar în conformitate cu prevederile prezentului Regulament.
61. Deciziile prevăzute la pct. 60 din prezentul Regulament sînt aduse la cunoştinţă fără întîrziere producătorului sau reprezentantului său autorizat, care va fi informat totodată cu privire la căile de atac pe care le are la dispoziţie, conform reglementărilor în vigoare, cît şi cu privire la termenul-limită pînă la care pot fi exercitate căile de atac.
62. În cazul unei decizii de natura celor prevăzute la pct. 60, părţile interesate sus-menţionate au posibilitatea de a-şi expune în prealabil punctul de vedere, cu excepţia cazului în care consultarea directă nu este posibilă, datorită urgenţei măsurilor ce urmează să fie adoptate.
Prevederile alineatului unu din prezentul punct referitor la asigurarea confidenţialităţii nu aduc atingere obligaţiilor Agenţiei şi ale organismelor recunoscute în ceea ce priveşte informarea reciprocă şi difuzarea avertismentelor şi nici obligaţiilor persoanelor care trebuie să furnizeze informaţii ce cad sub incidenţa legislaţiei penale.
64. Nu sînt considerate confidenţiale următoarele informaţii:
1) informaţiile privind înregistrarea persoanelor responsabile de introducerea pe piaţă a dispozitivelor, conform prevederilor pct. 40 din prezentul Regulament;
2) informaţiile destinate utilizatorilor transmise de producător, reprezentantul autorizat sau distribuitor în legătură cu o anumită măsură, potrivit prevederilor pct. 13 din prezentul Regulament;
3) informaţiile din certificatele emise, modificate, suplimentate, suspendate sau retrase.
66. Agenţia asigură cooperarea cu autorităţile competente din alte state privind domeniul dispozitivelor medicale implantabile active.
2. Dispozitivele asigură atingerea performanţelor prevăzute de către producător să fie proiectate şi, respectiv, fabricate astfel încît să îndeplinească una sau mai multe dintre funcţiile menţionate la pct. 2 din prezentul Regulament, conform celor specificate de producător.
3. Caracteristicile şi performanţele specificate la pct. 1 şi 2 din prezenta anexă nu trebuie să se deprecieze astfel încît să compromită starea clinică şi securitatea pacienţilor sau, după caz, a altor persoane pe întreaga durată de viaţă a dispozitivului estimată de producător, în condiţiile în care dispozitivul este supus unor solicitări posibile în condiţii normale de utilizare.
4. Dispozitivele urmează să fie proiectate, fabricate şi ambalate astfel încît caracteristicile şi performanţele acestora să nu fie afectate negativ ca urmare a condiţiilor de depozitare şi transport stabilite de producător (temperatură, umiditate etc.).
5. Orice efecte secundare sau stări nedorite trebuie să constituie riscuri acceptabile în raport cu performanţele pentru care este destinat dispozitivul.
6. Demonstrarea conformităţii cu cerinţele esenţiale trebuie să includă o evaluare clinică potrivit prevederilor prevăzute în anexa nr. 7 la prezentul Regulament.
8. Dispozitivele implantabile sînt proiectate, fabricate şi ambalate în ambalaje de unică folosinţă, conform procedurilor corespunzătoare, pentru a se asigura că acestea sînt sterile cînd sînt introduse pe piaţă, pe durata depozitării şi a transportului în condiţiile specificate de producător şi că rămîn sterile pînă cînd sînt dezambalate şi implantate.
9. Dispozitivele urmează să fie proiectate şi fabricate astfel încît să se elimine sau să se reducă cît mai mult posibil:
1) riscul de leziuni fizice, în legătură cu caracteristicile lor fizice, inclusiv cele dimensionale;
2) riscurile legate de utilizarea surselor de alimentare cu energie, îndeosebi atunci cînd se foloseşte electricitatea, cu referire, în mod particular, la izolaţie, curenţi de scurgere şi supraîncălzirea dispozitivelor;
3) riscurile legate de condiţiile de mediu previzibile în mod rezonabil, cum sînt cîmpurile magnetice, perturbaţiile electrice externe, descărcările electrostatice, presiunea sau variaţiile de presiune şi acceleraţie;
4) riscurile legate de tratamentele medicale, în particular cele care rezultă din utilizarea defibrilatoarelor sau a echipamentului electrochirurgical de înaltă frecvenţă;
5) riscurile legate de radiaţiile ionizante din substanţele radioactive incluse în dispozitiv;
6) riscurile care pot să apară în condiţiile în care nu sînt posibile întreţinerea şi calibrarea dispozitivelor, inclusiv:
a) creşterea excesivă a curenţilor de scurgere;
b) îmbătrînirea materialelor utilizate;
c) căldura excesivă generată de dispozitiv;
d) scăderea preciziei oricăror mecanisme de măsurare şi control.
10. Dispozitivele urmează să fie proiectate şi fabricate astfel încît să se garanteze caracteristicile şi performanţele cuprinse în capitolul I al prezentei anexe, cu o atenţie specială acordată pentru:
1) alegerea materialelor folosite, mai ales în ceea ce priveşte aspectele legate de toxicitate;
2) compatibilitatea mutuală dintre materialele utilizate şi ţesuturile biologice, celulele şi fluidele organismului, în concordanţă cu utilizarea preconizată a dispozitivului;
3) compatibilitatea dispozitivelor cu substanţele pe care sînt destinate să le administreze;
4) calitatea conexiunilor, în particular cu privire la securitate;
5) fiabilitatea sursei de energie;
6) protecţia împotriva scurgerilor, dacă este cazul;
7) funcţionarea corectă a sistemelor de programare şi control, inclusiv software-ul. În cazul dispozitivelor care încorporează software sau care sînt ele însele software medical, software-ul trebuie validat în conformitate cu nivelul tehnicii la momentul respectiv, luîndu-se în considerare principiile dezvoltării ciclului de viaţă, gestionării riscurilor, validării şi verificării.
11. În cazul în care un dispozitiv încorporează, ca parte integrantă, o substanţă care, dacă este folosită separat, se consideră ca fiind un medicament în sensul definiţiei prevăzute în legislaţia cu privire la medicamente şi care acţionează asupra organismului uman printr-o acţiune auxiliară celei a dispozitivului, calitatea, siguranţa şi utilitatea acelei substanţe se verifică prin analogie cu metodele specificate în normele şi protocoalele analitice, farmacotoxicologice şi clinice referitoare la testarea medicamentelor.
12. În cazul substanţelor menţionate la pct. 11 din prezenta anexă, organismul recunoscut, după ce a verificat utilitatea substanţei ca parte a dispozitivului medical şi ţinînd cont de scopul propus al dispozitivului, solicită avizul ştiinţific al Agenţiei sau al Agenţiei Europene pentru Medicamente (European Medicines Agency, în continuare – EMA), care hotărăşte cu privire la calitatea şi siguranţa substanţei, inclusiv raportul stabilit între beneficiile şi riscurile clinice ale încorporării substanţei în dispozitiv. La emiterea avizului, Agenţia sau EMA ia în considerare procesul de fabricaţie şi datele referitoare la utilitatea încorporării substanţei în dispozitiv, determinate de către organismul recunoscut.
13. În cazul în care un dispozitiv încorporează, ca parte integrantă, un derivat din sînge uman, organismul recunoscut, după ce a verificat utilitatea substanţei ca parte a dispozitivului medical şi ţinînd cont de scopul propus al dispozitivului, solicită avizul ştiinţific al Agenţiei sau al EMA, care hotărăşte cu privire la calitatea şi siguranţa substanţei, inclusiv raportul stabilit între beneficiile şi riscurile clinice ale încorporării derivatului din sînge uman în dispozitiv. La emiterea avizului, Agenţia sau EMA ia în considerare procesul de fabricaţie şi datele referitoare la utilitatea încorporării substanţei în dispozitiv, determinate de către organismul recunoscut.
14. În cazul în care se aduc modificări unei substanţe auxiliare încorporate într-un dispozitiv, în special dacă sînt legate de procesul de fabricaţie a acesteia, organismul recunoscut este informat cu privire la modificări şi consultă agenţia implicată în consultarea iniţială, pentru a confirma menţinerea gradului iniţial de calitate şi siguranţă al substanţei auxiliare. Agenţia ţine seama de datele referitoare la utilitatea încorporării substanţei în dispozitiv, determinate de organismul recunoscut, pentru a se asigura că modificările nu au un impact negativ asupra raportului stabilit între beneficiile şi riscurile adăugării substanţei în dispozitiv.
15. În cazul în care Agenţia, implicată în consultarea iniţială, a obţinut informaţii cu privire la substanţa auxiliară care ar putea avea impact asupra raportului stabilit între beneficiile şi riscurile adăugării substanţei în dispozitiv, aceasta consiliază organismului recunoscut, indiferent dacă informaţiile au sau nu impact asupra raportului stabilit între beneficiile şi riscurile adăugării substanţei în dispozitiv. Organismul recunoscut ţine seama de avizul ştiinţific actualizat şi reanalizează evaluarea sa din cadrul procedurii de evaluare a conformităţii.
16. Dispozitivele şi, după caz, părţile lor componente urmează a fi identificate pentru a permite luarea oricărei măsuri specificate în pct. 20 din prezenta anexă în caz de identificare a unui risc potenţial determinat de dispozitive sau de părţile lor componente.
17. Dispozitivele poartă un cod prin care acestea şi producătorul lor să poată fi identificate fără echivoc (în particular, cu privire la tipul dispozitivului şi anul fabricaţiei); acest cod asigură accesibilitatea pentru a fi citit, dacă este necesar, fără a fi nevoie de o intervenţie chirurgicală.
18. Dacă un dispozitiv sau accesoriile sale poartă instrucţiuni necesare pentru funcţionarea acestuia ori indică parametrii de funcţionare sau de reglare cu ajutorul unui sistem de vizualizare, atunci aceste informaţii asigură claritate pentru utilizator şi, dacă este cazul, pentru pacient.
19. Fiecare dispozitiv este însoţit de următoarele informaţii specifice, scrise lizibil şi care nu pot fi şterse, dacă este cazul, sub forma unor simboluri general recunoscute:
1) pe ambalajul steril:
a) metoda de sterilizare;
b) indicaţia care permite ca ambalajul să fie identificat ca atare;
c) numele şi adresa producătorului;
d) descrierea dispozitivului;
e) cuvintele „exclusiv pentru investigaţie clinică”, dacă dispozitivul este destinat investigaţiilor clinice;
f) cuvintele „dispozitiv fabricat la comandă”, dacă dispozitivul este fabricat la comandă;
g) declaraţia că dispozitivul implantabil este steril;
h) luna şi anul fabricaţiei;
i) indicaţia cu termenul pînă la care este posibilă implantarea dispozitivului în condiţii de securitate;
2) pe ambalajul de vînzare:
a) numele şi adresa producătorului şi a reprezentantului autorizat, în cazul în care producătorul nu are sediul social înregistrat în Republica Moldova;
b) descrierea dispozitivului;
c) scopul propus al dispozitivului;
d) caracteristicile relevante pentru utilizarea acestuia;
e) cuvintele „exclusiv pentru investigaţie clinică”, dacă dispozitivul este destinat investigaţiilor clinice;
f) cuvintele „dispozitiv fabricat la comandă”, dacă dispozitivul este fabricat la comandă;
g) declaraţia că dispozitivul implantabil este steril;
h) luna şi anul fabricaţiei;
i) indicaţia privind termenul pînă la care dispozitivul poate fi implantat în condiţii de securitate;
j) condiţiile de transport şi depozitare pentru dispozitiv;
k) în cazul unui dispozitiv prevăzut la pct. 5 din prezentul Regulament, indicaţia precum că dispozitivul conţine un derivat din sînge uman.
20. La introducerea pe piaţă, fiecare dispozitiv urmează a fi însoţit de:
1) instrucţiunile de utilizare în care să se indice următoarele date specifice:
a) anul autorizării pentru aplicarea marcajului de conformitate;
b) detaliile prevăzute la pct. 19 subpct. 1) şi 2), cu excepţia celor de la lit. h) şi i);
c) performanţele la care se face referire la pct. 2 din prezenta anexă, precum şi orice efecte secundare nedorite;
d) informaţiile care permit medicului să selecteze un dispozitiv adecvat şi software-ul corespunzător, precum şi accesoriile;
e) informaţiile care constituie instrucţiunile de utilizare şi care permit medicului şi, după caz, pacientului să utilizeze corect dispozitivul, software-ul şi accesoriile acestuia, precum şi informaţiile despre natura, domeniul şi periodicitatea verificărilor şi testelor de funcţionare şi, după caz, măsurile de întreţinere;
f) informaţiile care să permită, dacă este cazul, evitarea anumitor riscuri legate de implantarea dispozitivului;
g) informaţiile referitoare la riscurile unor interferenţe reciproce, legate de prezenţa dispozitivului în timpul unor investigaţii sau tratamente specifice; riscurile de interferenţe reciproce reprezintă efecte adverse asupra dispozitivului cauzate de instrumentele prezente la momentul investigaţiilor sau tratamentelor şi viceversa;
h) instrucţiunile necesare în eventualitatea deteriorării ambalajului steril şi, după caz, detaliile privind metodele adecvate de resterilizare;
i) indicaţia, unde este cazul, cu referire la posibilitatea reutilizării dispozitivului, cu condiţia că este recondiţionat sub responsabilitatea producătorului, pentru a corespunde cerinţelor esenţiale;
2) broşura cu instrucţiuni de utilizare trebuie să includă şi detalii care să permită medicului să instruiască pacientul cu privire la contraindicaţiile şi precauţiile necesare. Aceste detalii cuprind în special:
a) informaţiile care să permită stabilirea duratei de viaţă a sursei de energie;
b) precauţiile necesare în cazul în care apar modificări ale performanţelor dispozitivului;
c) precauţiile necesare privind expunerea, în condiţii de mediu previzibile în mod rezonabil, la cîmpuri magnetice, influenţe electrice exterioare, descărcări electrostatice, presiune sau variaţii de presiune, acceleraţie etc.;
d) informaţiile adecvate privind medicamentele pe care dispozitivul este destinat a le administra;
e) data emiterii sau a ultimei revizuiri a instrucţiunilor de utilizare.
21. Confirmarea că dispozitivul îndeplineşte cerinţele cu privire la caracteristicile şi performanţele prevăzute în capitolul I al prezentei anexe, în condiţii normale de funcţionare, şi că evaluarea efectelor secundare sau nedorite se realizează în baza datelor clinice, stabilite conform prevederilor din anexa nr. 7 la prezentul Regulament.
2. Declaraţia de conformitate este procedura prin intermediul căreia producătorul care îndeplineşte obligaţiile impuse la pct. 1 din prezenta anexă asigură şi declară că produsele în cauză îndeplinesc prevederile prezentului Regulament, care le sînt aplicabile.
3. Producătorul sau reprezentantul său autorizat cu sediul în Republica Moldova aplică marcajul de conformitate, potrivit prevederilor capitolului X din prezentul Regulament, şi întocmeşte, în scris, o declaraţie de conformitate. Această declaraţie se referă la unul sau mai multe dispozitive identificate clar prin intermediul denumirii sau al codului produsului ori al altei referinţe lipsite de ambiguitate şi se păstrează de către producător. Marcajul de conformitate este însoţit de numărul de identificare al organismului recunoscut responsabil.
Cererea include:
a) toate informaţiile adecvate referitoare la categoria de produse a căror fabricaţie este vizată;
b) documentaţia referitoare la sistemul calităţii;
c) angajamentul producătorului de a îndeplini obligaţiile ce decurg din sistemul calităţii aprobat;
d) angajamentul producătorului de a menţine sistemul calităţii aprobat în aşa fel încît acesta să rămînă adecvat şi eficace;
e) angajamentul producătorului de a institui şi de a menţine actualizat un sistem de supraveghere postvînzare, care să includă dispoziţiile menţionate în anexa nr. 7 la prezentul Regulament. Acest angajament include obligaţia asumată de către producător de a comunica autorităţilor competente următoarele incidente imediat ce a luat cunoştinţă despre acestea:
(i) orice deteriorare a caracteristicilor sau performanţelor dispozitivului;
(ii) orice inexactitate din instrucţiunile de utilizare care ar putea conduce ori au condus la decesul unui pacient sau la deteriorarea stării de sănătate a acestuia;
(iii) orice argument tehnic sau medical care are ca rezultat retragerea de pe piaţă a unui dispozitiv de către producător.
5. Aplicarea sistemului calităţii asigură conformitatea dispozitivelor cu prevederile prezentului Regulament, care le sînt aplicabile, în toate etapele, de la proiectare pînă la controalele finale.
6. Toate elementele, cerinţele şi dispoziţiile adoptate de producător pentru sistemul calităţii sînt documentate în mod sistematic şi ordonat, sub formă de proceduri şi declaraţii scrise privind politica de calitate. Această documentaţie a sistemului calităţii asigură posibilitatea interpretării uniforme a politicilor calităţii şi a procedurilor, cum ar fi: programe ale calităţii, planuri ale calităţii, manuale ale calităţii şi înregistrări ale calităţii. Aceasta include, în special, documentele, datele şi înregistrările corespunzătoare generate de procedurile menţionate la lit.c) din prezentul punct.
Documentaţia include, în special, o descriere adecvată a:
a) obiectivelor producătorului privind calitatea;
b) organizării afacerilor şi, în special:
(i) a structurilor organizatorice, a responsabilităţilor personalului de conducere şi a autorităţii acesteia în legătură cu organizarea, în cazul în care este vizată calitatea proiectării şi fabricaţia produselor;
(ii) a metodelor de monitorizare a funcţionării eficiente a sistemului calităţii şi, în special, capacitatea acestuia de a determina calitatea dorită a proiectului şi a produselor, inclusiv controlul produselor care nu sînt conforme;
(iii) în cazul în care proiectarea, fabricarea şi/sau inspecţia şi testarea finală a produselor sau a elementelor acestora sînt efectuate de o terţă parte, a metodelor de monitorizare a funcţionării eficace a sistemului calităţii şi, în special, tipul şi amploarea controalelor aplicate terţei părţi în cauză;
c) procedurilor de monitorizare şi verificare a proiectelor produselor şi, în special:
(i) a specificaţiilor de proiectare, inclusiv a standardelor care se aplică, şi o descriere a soluţiilor adoptate pentru a îndeplini cerinţele esenţiale aplicabile produselor, atunci cînd standardele menţionate la pct. 17 din prezentul Regulament nu sînt aplicate integral;
(ii) a tehnicilor de control şi verificare a proiectării, a proceselor şi acţiunilor sistematice ce urmează a fi utilizate în cadrul proiectării produselor;
(iii) a declaraţiei care indică dacă dispozitivul cuprinde sau nu, ca parte integrantă, o substanţă ori un derivat din sînge uman menţionată/menţionat la pct.11-15 din anexa nr. 1 la prezentul Regulament, precum şi datele referitoare la testele efectuate în această privinţă, necesare pentru a evalua securitatea, calitatea şi utilitatea acelei substanţe sau ale derivatului din sînge uman în cauză, conform scopului propus al dispozitivului;
(iv) a evaluării preclinice;
(v) a evaluării clinice menţionate în anexa nr. 7 la prezentul Regulament;
d) tehnicilor de control şi de asigurare a calităţii în faza de fabricaţie şi, în special:
(i) a proceselor şi procedurilor ce urmează a fi utilizate, în special cu privire la sterilizare, achiziţie de materiale şi la documentele relevante;
(ii) a procedurilor de identificare a produsului, întocmite şi actualizate din desene, specificaţii sau alte documente relevante, în fiecare fază a procesului de fabricaţie;
e) testelor şi verificărilor adecvate, care vor fi efectuate înainte, în timpul şi după procesul de fabricaţie, a frecvenţei acestora şi a echipamentului de testare folosit.
7. Fără a aduce atingere dispoziţiilor pct. 60, 61 şi 62 din prezentul Regulament, organismul recunoscut efectuează un audit al sistemului calităţii, pentru a stabili dacă acesta îndeplineşte cerinţele menţionate la pct. 5 şi 6 la prezenta anexă. Sistemele calităţii care folosesc standardele armonizate corespunzătoare sînt conforme cerinţelor sus-menţionate.
Echipa responsabilă de evaluare include cel puţin un membru care are experienţă în evaluarea tehnologiilor respective.
Procedura de evaluare include o inspecţie la sediul producătorului şi, în cazuri justificate în mod corespunzător, la sediile furnizorilor producătorului şi/sau ale subcontractanţilor, pentru a inspecta procesele de fabricaţie.
Decizia se comunică producătorului după inspecţia finală. Aceasta cuprinde concluziile activităţii de control şi o evaluare argumentată.
8. Producătorul informează organismul recunoscut, care a aprobat sistemul calităţii, cu privire la orice plan de modificare a sistemului calităţii. Organismul recunoscut evaluează modificările propuse şi verifică dacă sistemul calităţii astfel modificat mai respectă cerinţele menţionate la pct. 5 şi 6 din prezenta anexă; acesta face cunoscută decizia sa producătorului. Această decizie cuprinde concluziile activităţii de control şi o evaluare argumentată.
10. Cererea descrie proiectarea, procesul de fabricaţie şi performanţele produsului respectiv şi include documentele necesare evaluării conformităţii produsului cu cerinţele prezentului Regulament, în special, cu cele prevăzute la pct. 5 şi pct.6 subpct.1) lit.c) şi d) din prezenta anexă.
Cererea include printre altele:
a) specificaţiile de proiectare, inclusiv standardele care au fost aplicate;
b) dovada din care să rezulte că standardele respective sînt corespunzătoare, mai ales în cazul în care standardele menţionate la pct. 17 din prezentul Regulament nu au fost aplicate integral. Această dovadă urmează să includă rezultatele încercărilor adecvate efectuate de către producător sau sub responsabilitatea acestuia;
c) declaraţia din care să rezulte dacă dispozitivul încorporează sau nu, ca parte integrantă, o substanţă de tipul celor menţionate la pct.11-15 din anexa nr.1 la prezentul Regulament, a cărei acţiune în asociere cu dispozitivul poate determina biodisponibilitatea sa, însoţită de date asupra investigaţiilor relevante realizate;
d) evaluarea clinică menţionată în anexa nr. 7 la prezentul Regulament;
e) proiectul broşurii cu instrucţiunile de utilizare.
11. Organismul recunoscut examinează solicitarea, iar dacă produsul este în conformitate cu prevederile relevante ale Regulamentului, eliberează solicitantului un certificat de examinare a proiectului. Organismul recunoscut poate cere ca solicitarea să fie completată cu teste sau cu probe suplimentare care să permită evaluarea conformităţii cu cerinţele prezentului Regulament. Certificatul cuprinde concluziile examinării, condiţiile de validitate ale acestuia, datele necesare identificării proiectului aprobat şi, după caz, descrierea scopului propus al produsului.
12. În cazul dispozitivelor menţionate la pct. 12 din anexa nr. 1 la prezentul Regulament, înainte de a lua o decizie, organismul recunoscut consultă, în ceea ce priveşte aspectele vizate la acel punct, Agenţia sau EMA. Avizul Agenţiei sau al EMA se emite în termen de 210 zile de la primirea unei documentaţii valide. Avizul ştiinţific al Agenţiei sau al EMA trebuie să fie inclus în documentaţia privind dispozitivul. La luarea deciziei, organismul recunoscut ia în considerare punctele de vedere exprimate cu ocazia acestei consultări. Acesta transmite decizia sa finală organismului competent implicat.
13. În cazul dispozitivelor menţionate în capitolul II şi pct.13 din anexa nr. 1 la prezentul Regulament, avizul ştiinţific al Agenţiei sau al EMA trebuie inclus în documentaţia privind dispozitivul. Avizul se emite în termen de 210 zile de la primirea unei documentaţii valide. La luarea deciziei, organismul recunoscut ia în considerare avizul Agenţiei sau al EMA. Organismul recunoscut poate să nu elibereze certificatul, în cazul în care avizul ştiinţific al Agenţiei sau al EMA este nefavorabil. Acesta transmite decizia sa finală către Agenţie sau EMA.
14. Solicitantul informează organismul recunoscut care a emis certificatul de examinare a proiectului despre orice modificare efectuată la proiectul aprobat. Pentru modificările efectuate la proiectul aprobat este necesară obţinerea unei aprobări suplimentare de la organismul recunoscut care a emis certificatul de examinare a proiectului, în cazul în care aceste modificări pot afecta conformitatea cu cerinţele esenţiale din prezentul Regulament sau condiţiile prescrise pentru utilizarea produsului. Această aprobare suplimentară se acordă sub forma unui addendum la certificatul de examinare a proiectului.
16. Producătorul autorizează organismul recunoscut să efectueze toate inspecţiile necesare şi îi furnizează toate informaţiile, în special în legătură cu:
a) documentaţia privind sistemul calităţii;
b) datele prevăzute în acea parte a sistemului calităţii care se referă la proiect, cum ar fi rezultatele analizelor, calculelor, testelor, evaluarea preclinică, evaluarea clinică, planul de monitorizare clinică postvînzare şi rezultatele monitorizării clinice postvînzare, dacă este cazul;
c) datele prevăzute în partea sistemului calităţii care se referă la fabricaţie, cum ar fi: rapoartele referitoare la inspecţii, testele, standardizările/calibrările şi calificările personalului implicat.
17. Organismul recunoscut efectuează periodic inspecţiile şi evaluările necesare pentru a se asigura că producătorul aplică sistemul calităţii aprobat şi pune la dispoziţia producătorului un raport de evaluare.
18. În plus, organismul recunoscut poate face vizite neanunţate producătorului, punîndu-i acestuia la dispoziţie un raport de inspecţie.
a) declaraţia de conformitate;
b) documentaţia prevăzută la pct. 4 lit. b), în special documentaţia, datele şi înregistrările menţionate în pct. 5 şi 6 din prezenta anexă;
c) amendamentele specificate la pct.8 din prezenta anexă;
d) documentaţia specificată la pct. 10 din prezenta anexă;
e) deciziile şi rapoartele organismului recunoscut, specificate la pct. 8, 11, 12, 13, 17 şi 18 din prezenta anexă.
20. La cerere, organismul recunoscut pune la dispoziţia celorlalte organisme recunoscute şi a Agenţiei toate datele relevante cu privire la aprobările sistemelor calităţii emise, respinse sau retrase.
21. După fabricarea fiecărui lot de dispozitive prevăzute la pct. 5 din prezentul Regulament, producătorul informează organismul recunoscut despre eliberarea lotului de dispozitive şi îi transmite certificatul oficial de eliberare a lotului de derivat din sînge uman utilizat în dispozitiv, certificat emis de un laborator de stat.
2. Cererea pentru examinarea CE de tip se adresează de către producător sau de către reprezentantul său autorizat stabilit în Republica Moldova unui organism recunoscut.
Cererea cuprinde:
1) denumirea şi adresa producătorului şi a reprezentantului autorizat, dacă cererea este adresată de acesta din urmă;
2) declaraţia scrisă prin care se specifică faptul că o astfel de cerere nu a mai fost adresată nici unui alt organism recunoscut;
3) documentaţia descrisă la pct. 3 din prezenta anexă necesară pentru a permite evaluarea conformităţii unui eşantion reprezentativ din producţia respectivă (în continuare – tip), conform cerinţelor esenţiale din prezentul Regulament. Solicitantul asigură punerea unui tip la dispoziţia organismului recunoscut, iar acesta poate cere şi alte eşantioane, dacă este necesar.
3. Documentaţia urmează să asigure înţelegerea proiectării, fabricaţiei şi performanţelor dispozitivului şi cuprinde, în special, următoarele aspecte:
1) descrierea generală a tipului, inclusiv variantele avute în vedere, şi a utilizării (utilizărilor) prevăzute ale acestuia;
2) desenele de proiect, metodele de fabricaţie planificate, în special cu privire la sterilizare, precum şi diagramele de componente, subansambluri şi circuite;
3) descrierile şi explicaţiile necesare pentru înţelegerea desenelor şi diagramelor menţionate la subpct.2) din prezentul punct, precum şi descrierea funcţionării produsului;
4) lista standardelor menţionate la pct. 17 din prezentul Regulament, aplicate integral sau parţial, precum şi o descriere a soluţiilor adoptate pentru îndeplinirea cerinţelor esenţiale în cazul în care nu sînt aplicate standardele menţionate la pct. 17 din prezentul Regulament;
5) rezultatele calculelor de proiectare, analiza riscurilor, investigaţiile, testele tehnice efectuate;
6) declaraţia care indică dacă dispozitivul încorporează sau nu, ca parte integrantă, o substanţă ori un derivat din sînge uman, conform prevederilor de la pct.11-15 din anexa nr. 1 la prezentul Regulament, precum şi datele referitoare la testele efectuate în această privinţă, necesare pentru a evalua securitatea, calitatea şi utilitatea substanţei sau a derivatului din sînge uman, conform scopului propus al dispozitivului;
7) evaluarea preclinică;
8) evaluarea clinică menţionată în anexa nr. 7 la prezentul Regulament;
9) proiectul broşurii cu instrucţiunile de utilizare.
1) examinează şi evaluează documentaţia şi verifică dacă tipul este fabricat în conformitate cu această documentaţie; de asemenea, înregistrează elementele care au fost proiectate în conformitate cu prevederile aplicabile ale standardelor menţionate la pct. 17 din prezentul Regulament, precum şi elementele pentru care proiectarea nu se bazează pe prevederile acestor standarde;
2) efectuează sau solicită efectuarea inspecţiilor şi a încercărilor necesare pentru a verifica dacă soluţiile adoptate de producător îndeplinesc cerinţele esenţiale din prezentul Regulament, în cazul în care standardele menţionate la pct. 17 din prezentul Regulament nu au fost aplicate;
3) efectuează sau solicită efectuarea inspecţiilor şi a încercărilor necesare pentru a verifica dacă, în cazul în care producătorul a optat pentru aplicarea standardelor relevante, acestea au fost într-adevăr aplicate;
4) stabileşte de comun acord cu solicitantul locul unde se efectuează inspecţiile şi încercările necesare.
5. În cazul în care tipul este conform cu prevederile prezentului Regulament, organismul recunoscut eliberează solicitantului un certificat de examinare CE de tip. Certificatul cuprinde denumirea şi adresa producătorului, concluziile controlului, condiţiile de validitate şi datele necesare pentru identificarea tipului aprobat. Părţile relevante ale documentaţiei se anexează certificatului, iar o copie se păstrează de organismul recunoscut.
6. În cazul dispozitivelor menţionate la pct. 12 din anexa nr. 1 la prezentul Regulament, înainte de a lua o decizie, organismul recunoscut consultă Agenţia sau EMA în ceea ce priveşte aspectele vizate la acel punct.
Avizul Agenţiei sau al EMA se emite în termen de 210 zile de la primirea unei documentaţii valide. Avizul ştiinţific al Agenţiei sau al EMA trebuie inclus în documentaţia privind dispozitivul.
La luarea deciziei, organismul recunoscut ia în considerare punctele de vedere exprimate în aviz. Acesta transmite decizia sa finală organismului competent implicat.
7. În cazul dispozitivelor menţionate la pct.13 din anexa nr. 1 la Regulament, avizul ştiinţific al Agenţiei sau al EMA este inclus în documentaţia privind dispozitivul.
Avizul se emite în termen de 210 zile de la primirea unei documentaţii valide. La luarea deciziei, organismul recunoscut ia în considerare avizul Agenţiei sau al EMA.
Organismul recunoscut nu eliberează certificatul în cazul în care avizul ştiinţific al Agenţiei sau al EMA este nefavorabil. Acesta transmite decizia sa finală către Agenţie sau al EMA.
8. Solicitantul informează organismul recunoscut care a emis certificatul de examinare CE de tip cu privire la orice modificare efectuată asupra produsului aprobat.
9. Modificările asupra produsului aprobat trebuie să primească o altă aprobare din partea organismului recunoscut care a emis certificatul de examinare CE de tip, în cazul în care astfel de modificări pot afecta conformitatea cu cerinţele esenţiale sau condiţiile prevăzute pentru utilizarea produsului. Noua aprobare este eliberată, dacă este cazul, sub forma unui supliment la certificatul de examinare CE de tip iniţial.
11. Alte organisme recunoscute pot obţine o copie de pe certificatele de examinare CE de tip şi/sau de pe suplimentele acestora. Anexele la certificate trebuie să fie accesibile pentru alte organisme recunoscute, la solicitarea justificată a acestora şi după informarea prealabilă a producătorului.
12. Producătorul sau reprezentantul său autorizat păstrează documentaţia tehnică şi copiile certificatelor de examinare CE de tip şi suplimentelor acestora pentru o perioadă de cel puţin 15 ani de la fabricarea ultimului produs.
2. Producătorul sau reprezentantul său autorizat cu sediul în Republica Moldova ia măsurile necesare pentru ca procesul de fabricaţie să asigure conformitatea produselor cu tipul descris în certificatul de examinare CE de tip şi cu cerinţele aplicabile din prezentul Regulament. Producătorul sau reprezentantul său autorizat cu sediul în Republica Moldova aplică marcajul de conformitate pe fiecare produs şi emite, în scris, o declaraţie de conformitate.
3. Înainte de începerea fabricaţiei, producătorul redactează documentele care definesc procesul de fabricaţie, în special privind sterilizarea, împreună cu toate prevederile de rutină prestabilite, pentru a asigura o producţie omogenă şi conformitatea produselor cu tipul descris în certificatul de examinare CE de tip şi cu cerinţele relevante din prezentul Regulament.
4. Producătorul se angajează să instituie şi să menţină la zi un sistem de supraveghere postvînzare, care să includă dispoziţiile menţionate în anexa nr. 7 la prezentul Regulament. Acest angajament include obligaţia producătorului de a comunica Agenţiei, imediat ce a luat cunoştinţă de acestea, următoarele evenimente:
1) orice deteriorare a caracteristicilor sau performanţelor dispozitivului, precum şi orice inexactitate din instrucţiunile de utilizare, care ar putea conduce sau au condus la decesul unui pacient ori la deteriorarea stării de sănătate al acestuia;
2) orice argument tehnic sau medical care a condus la retragerea de pe piaţă a unui dispozitiv de către producător.
5. Organismul recunoscut efectuează examinările şi încercările necesare pentru a verifica dacă produsul este conform cu cerinţele prezentului Regulament, prin examinarea şi testarea statistică a produselor, în conformitate cu prevederile pct. 6 din prezenta anexă. Producătorul autorizează organismul recunoscut să evalueze eficienţa măsurilor adoptate, potrivit prevederilor pct. 3 din prezenta anexă, prin audit, unde este cazul.
6. Verificarea statistică
1) Producătorii prezintă produsele fabricate sub formă de loturi omogene şi iau toate măsurile necesare astfel încît procesul de fabricaţie să asigure uniformitatea fiecărui lot de producţie realizat.
2) Se prelevă aleatoriu un eşantion din fiecare lot. Produsele din cadrul eşantionului se examinează individual şi se efectuează încercările corespunzătoare, definite în standardele menţionate la pct. 17 din prezentul Regulament, sau încercările echivalente pentru verificarea conformităţii produselor cu modelul descris în certificatul de examinare CE de tip, în scopul deciziei acceptării sau refuzării lotului de producţie.
3) Controlul statistic al produselor se bazează pe atribute şi/sau variabile, ceea ce implică un sistem de prelevare a eşantioanelor, cu caracteristici operaţionale, care să asigure un nivel ridicat de securitate şi performanţă în funcţie de nivelul tehnicii la momentul respectiv. Sistemele de prelevare a eşantioanelor se stabilesc în conformitate cu standardele armonizate menţionate la pct. 17 din prezentul Regulament, luînd în considerare specificul categoriilor de produse în discuţie.
4) În cazul în care loturile sînt acceptate, organismul recunoscut aplică sau impune aplicarea numărului său de identificare pe fiecare produs şi eliberează un certificat de conformitate privind încercările efectuate. Toate produsele din lot pot fi introduse pe piaţă, cu excepţia produselor neconforme din eşantionul examinat.
În cazul în care un lot este respins, organismul recunoscut ia măsurile necesare pentru a preveni introducerea pe piaţă a lotului respectiv. În eventualitatea respingerii frecvente a loturilor, organismul recunoscut suspendă verificarea statistică.
Producătorul poate, sub responsabilitatea organismului recunoscut, să aplice numărul de identificare al acestuia în timpul procesului de fabricaţie.
5) Producătorul sau reprezentantul său autorizat trebuie să asigure că este capabil de a furniza, la cerere, certificatele de conformitate emise de organismul recunoscut.
7. După fabricarea fiecărui lot de dispozitive prevăzute la pct. 5 din prezentul Regulament, producătorul informează organismul recunoscut despre eliberarea lotului de dispozitive şi îi transmite certificatul oficial de eliberare a lotului de derivat din sînge uman utilizat în dispozitiv, certificat emis de un laborator de stat.
2. Declaraţia de conformitate este o parte a procedurii prin care producătorul care îndeplineşte obligaţiile impuse la pct. 1 din prezenta anexă garantează şi declară că produsele respective sînt conforme cu tipul descris în certificatul de examinare CE de tip şi respectă prevederile prezentului Regulament, aplicabile acestora.
Producătorul sau reprezentantul său autorizat cu sediul în Republica Moldova aplică marcajul de conformitate potrivit prevederilor capitolului X din prezentul Regulament şi întocmeşte, în scris, o declaraţie de conformitate. Această declaraţie acoperă unul sau mai multe dispozitive identificate clar prin intermediul denumirii ori al codului produsului sau al altei referinţe lipsite de ambiguitate şi se păstrează de către producător. Marcajul de conformitate este însoţit de numărul de identificare al organismului recunoscut responsabil.
Cererea cuprinde:
a) toate informaţiile adecvate referitoare la produsele pe care intenţionează să le fabrice;
b) documentaţia sistemului calităţii;
c) angajamentul producătorului de a îndeplini obligaţiile ce decurg din sistemul calităţii aprobat;
d) angajamentul producătorului de a menţine sistemul calităţii aprobat în aşa fel încît acesta să rămînă adecvat şi eficace;
e) documentaţia tehnică privind tipul aprobat şi o copie a certificatului de examinare CE de tip, după caz;
f) angajamentul producătorului de a institui şi de a menţine actualizat un sistem de supraveghere postvînzare, care să includă dispoziţiile prevăzute în anexa nr. 7 la prezentul Regulament. Acest angajament include obligaţia asumată de către producător de a comunica Agenţiei următoarele incidente imediat ce a luat cunoştinţă despre acestea:
(i) orice deteriorare a caracteristicilor sau performanţelor dispozitivului, precum şi orice inexactitate din instrucţiunile de utilizare, care ar putea duce sau au dus la decesul unui pacient ori la deteriorarea stării sale de sănătate;
(ii) orice argument tehnic sau medical care a condus la retragerea unui dispozitiv de pe piaţă de către producător.
4. Aplicarea sistemului calităţii asigură că produsele sînt conforme cu tipul descris în certificatul de examinare CE de tip.
5. Toate elementele, cerinţele şi dispoziţiile adoptate de producător pentru sistemul calităţii sînt documentate în mod sistematic şi ordonat sub formă de proceduri şi politici scrise. Această documentaţie a sistemului calităţii trebuie să permită o interpretare uniformă a politicilor calităţii şi a procedurilor, cum ar fi: programele calităţii, planurile calităţii, manualele calităţii şi înregistrările calităţii.
Documentaţia include, în special, o descriere adecvată a:
a) obiectivelor producătorului privind calitatea;
b) organizării afacerilor, în special:
(i) a structurilor organizatorice, responsabilităţilor personalului de conducere şi a autorităţii acestuia, în cazul în care este vizată fabricaţia produselor;
(ii) a metodelor de monitorizare a funcţionării eficiente a sistemului calităţii, în special capacitatea acestuia de a determina calitatea dorită a produselor, inclusiv controlul produselor care nu sînt conforme;
(iii) în cazul în care fabricarea şi/sau inspecţia şi testarea finală a produselor sau a elementor acestora sînt efectuate de o terţă parte, a metodelor de monitorizare a funcţionării eficace a sistemului calităţii, în special tipul şi amploarea controalelor aplicate terţei părţi în cauză;
c) tehnicilor de control şi de asigurare a calităţii în faza de fabricaţie, în special:
(i) a proceselor şi procedurilor ce urmează a fi utilizate, în special, cu privire la sterilizare, achiziţie de materiale şi documentele relevante;
(ii) a procedurilor de identificare a produsului întocmite şi actualizate din desene, specificaţii sau alte documente relevante, în fiecare fază a procesului de fabricaţie;
d) testelor şi verificărilor adecvate, efectuate înainte, în timpul şi după procesul de fabricaţie, a frecvenţei acestora şi a echipamentului de testare folosit.
6. Fără a aduce atingere dispoziţiilor pct. 58 şi 59 din prezentul Regulament, organismul recunoscut efectuează un audit al sistemului calităţii pentru a stabili dacă acesta îndeplineşte cerinţele menţionate la pct. 4 şi 5 din prezenta anexă. Se consideră că sistemele calităţii care folosesc standardele armonizate corespunzătoare sînt conforme acestor cerinţe.
Echipa responsabilă de evaluare include cel puţin un membru care are experienţă în evaluarea tehnologiilor respective. Procedura de evaluare include o inspecţie la sediul producătorului.
Decizia se comunică producătorului după inspecţia finală. Aceasta cuprinde concluziile activităţii de control şi o evaluare motivată.
7. Producătorul informează organismul recunoscut care a aprobat sistemul calităţii cu privire la orice plan de modificare a sistemului calităţii. Organismul recunoscut evaluează modificările propuse şi verifică dacă sistemul calităţii astfel modificat mai respectă cerinţele menţionate la pct. 4 şi 5 din prezenta anexă; acesta face cunoscută decizia sa producătorului. Această decizie cuprinde concluziile activităţii de control şi o evaluare argumentată.
9. Producătorul autorizează organismul recunoscut să efectueze toate inspecţiile necesare şi îi furnizează toate informaţiile necesare, în special în legătură cu:
a) documentaţia privind sistemul calităţii;
b) documentaţia tehnică;
c) datele prevăzute în sistemul calităţii referitoare la fabricaţie, cum ar fi: rapoartele de inspecţie, testele, standardizarea/calibrarea şi calificările personalului
implicat etc.
10. Organismul recunoscut efectuează periodic inspecţiile şi evaluările necesare pentru a se asigura că producătorul aplică sistemul calităţii aprobat şi pune la dispoziţia producătorului un raport de evaluare.
11. În plus, organismul recunoscut, dacă este necesar, face vizite neanunţate producătorului, punîndu-i acestuia la dispoziţie un raport de inspecţie.
12. Organismul recunoscut comunică celorlalte organisme recunoscute toate datele relevante referitoare la aprobările privind sistemele de calitate emise, respinse sau retrase.
13. După fabricarea fiecărui lot de dispozitive prevăzute la pct. 5 din prezentul Regulament, producătorul informează organismul recunoscut despre eliberarea lotului de dispozitive şi îi transmite certificatul oficial de eliberare a lotului de derivat din sînge uman utilizat în dispozitiv, certificat emis de un laborator de stat sau de un laborator desemnat de stat.
2. Declaraţia cuprinde următoarele informaţii:
1) pentru dispozitivele fabricate la comandă:
a) denumirea şi adresa producătorului;
b) informaţiile necesare pentru identificarea produsului în cauză;
c) o declaraţie din care să rezulte că dispozitivul este destinat pentru a fi utilizat în exclusivitate de către un anumit pacient, menţionîndu-se numele pacientului;
d) numele practicianului medical calificat în mod corespunzător care a făcut prescripţia şi, după caz, numele clinicii implicate;
e) caracteristicile specifice ale produsului prezentate de prescripţia medicală;
f) o declaraţie din care să rezulte că dispozitivul respectiv satisface cerinţele esenţiale prevăzute în anexa nr. 1 la prezentul Regulament şi care, după caz, să indice care dintre cerinţele esenţiale nu au fost îndeplinite integral, împreună cu motivaţia;
2) pentru dispozitivele destinate investigaţiilor clinice prevăzute în anexa nr. 7 la prezentul Regulament:
a) datele care să permită identificarea dispozitivelor respective;
b) planul investigaţiei clinice;
c) broşura investigatorului;
d) confirmarea asigurării subiecţilor;
e) documentele utilizate pentru obţinerea consimţămîntului în cunoştinţă de cauză;
f) o declaraţie care indică dacă dispozitivul încorporează sau nu, ca parte integrantă, o substanţă ori un derivat din sînge uman prevăzută/prevăzut la pct. 11-15 din anexa nr. 1 la prezentul Regulament;
g) avizul comisiei de etică implicate şi detaliile privind aspectele abordate în avizul său;
h) numele practicianului medical calificat în mod corespunzător sau al altei persoane autorizate şi al instituţiei responsabile pentru investigaţii;
i) locul şi data începerii investigaţiilor şi durata programată pentru acestea;
j) declaraţia din care să rezulte că dispozitivul respectiv este conform cu cerinţele esenţiale, prevăzute în anexa nr. 1 la prezentul Regulament, excluzînd aspectele ce constituie obiectul investigaţiilor, şi că, în ceea ce priveşte aceste aspecte, s-au luat toate măsurile de precauţie pentru a proteja sănătatea şi securitatea pacientului.
3. Producătorul se angajează să pună la dispoziţia Agenţiei următoarele:
1) pentru dispozitivele fabricate la comandă, documentaţia care indică amplasamentul (amplasamentele) de producţie şi care permite înţelegerea proiectului, a fabricaţiei şi a performanţelor produsului, inclusiv performanţele urmărite, pentru a permite evaluarea conformităţii cu cerinţele prezentului Regulament.
Producătorul ia toate măsurile necesare pentru ca procesul de fabricaţie să asigure că produsele fabricate sînt conforme cu documentaţia menţionată mai sus;
2) pentru dispozitivele destinate investigaţiilor clinice, documentaţia cuprinde, de asemenea:
a) descrierea generală a produsului şi a utilizărilor prevăzute;
b) desenele de proiect, metodele de fabricaţie, în special în ceea ce priveşte sterilizarea, şi diagramele de componente, subansamblurile, circuitele;
c) descrierile şi explicaţiile necesare pentru înţelegerea respectivelor desene şi diagrame şi a funcţionării produsului;
d) rezultatele analizei riscurilor şi o listă cuprinzînd standardele prevăzute la pct. 17 din prezentul Regulament, aplicate integral sau în parte, precum şi o descriere a soluţiilor adoptate pentru îndeplinirea cerinţelor esenţiale din prezentul Regulament, în cazul în care standardele menţionate la pct. 17 din prezentul Regulament nu au fost aplicate;
e) în cazul în care dispozitivul încorporează, ca parte integrantă, o substanţă sau un derivat din sînge uman menţionată/menţionat la pct. 11-15 din anexa nr. 1 la prezentul Regulament, datele referitoare la testele efectuate în această privinţă, necesare pentru a evalua securitatea, calitatea şi utilitatea substanţei sau a derivatului de sînge uman, conform scopului propus al dispozitivului;
f) rezultatele calculelor proiectului, verificărilor şi testelor tehnice executate.
Producătorul ia toate măsurile necesare pentru ca procesul de fabricaţie să asigure că produsele fabricate sînt în conformitate cu documentaţia menţionată la pct. 3 subpct. 1) şi subpct. 2) lit. a)-f) din prezenta anexă.
Producătorul, dacă este necesar, poate autoriza evaluarea eficienţei acestor măsuri prin audit.
4. Informaţiile incluse în declaraţiile care intră sub incidenţa prezentei anexe se păstrează pentru o perioadă de cel puţin 15 ani de la data fabricaţiei ultimului produs.
5. Pentru dispozitivele fabricate la comandă, producătorul se angajează să revizuiască şi să documenteze experienţa acumulată după încheierea fazei de producţie, inclusiv dispoziţiile menţionate în anexa nr. 7 la prezentul Regulament, şi să implementeze mijloacele adecvate pentru aplicarea oricăror măsuri corective necesare. Acest angajament include obligaţia producătorului de a informa autorităţile competente cu privire la următoarele incidente imediat ce a aflat de existenţa lor, precum şi la măsurile corective relevante:
1) orice funcţionare defectuoasă sau deteriorare a caracteristicilor şi/sau a performanţelor unui dispozitiv, precum şi orice caz de inadecvare a etichetării sau a instrucţiunilor de utilizare, care pot să conducă sau au condus la decesul ori la deteriorarea severă a stării de sănătate a unui pacient sau utilizator;
2) orice cauză de ordin tehnic sau medical legată de caracteristicile sau performanţele unui dispozitiv, care, din motivele menţionate la subp.1) din prezentul punct, conduce la retragerea sistematică de pe piaţă de către producător a dispozitivelor de acelaşi tip.
a) fie o evaluare critică a literaturii ştiinţifice, curent disponibile, cu privire la securitatea, performanţele, caracteristicile proiectului şi scopul propus al dispozitivului, în care:
(i) se demonstrează echivalenţa dispozitivului cu dispozitivul la care fac referire datele; şi
(ii) datele demonstrează în mod adecvat conformitatea cu cerinţele esenţiale relevante;
b) fie o evaluare critică a rezultatelor tuturor investigaţiilor clinice efectuate;
c) fie o evaluare critică a datelor clinice combinate prevăzute la lit. a şi b din prezentul subpunct.
2. Se efectuează investigaţii clinice, cu excepţia cazurilor în care se justifică utilizarea datelor clinice existente.
3. Pentru evaluarea clinică şi rezultatul său se prezintă documente justificative. Documentaţia tehnică a dispozitivului include şi/sau face trimitere la documentaţia clinică în cauză.
4. Evaluarea clinică şi documentaţia aferentă se actualizează activ cu datele obţinute în cursul supravegherii după introducerea pe piaţă. În cazul în care se constată că supravegherea clinică după introducerea pe piaţă, ca parte integrantă a planului de supraveghere a dispozitivului după introducerea pe piaţă, nu este necesară, acest lucru se justifică şi se documentează în mod adecvat.
5. În cazul în care se constată că demonstrarea conformităţii cu cerinţele esenţiale pe baza datelor clinice nu este adecvată, se furnizează o justificare corespunzătoare a acestei excluderi, pe baza rezultatelor gestionării riscurilor şi luînd în considerare caracteristicile specifice ale interacţiunii dintre dispozitiv şi organismul uman, performanţele clinice prevăzute şi cererile producătorului. În cazul în care demonstrarea conformităţii cu cerinţele esenţiale se bazează exclusiv pe evaluarea performanţelor, teste pe banc şi evaluare preclinică, este necesar să se demonstreze în mod corespunzător că această metodă este adecvată.
6. Toate datele rămîn confidenţiale, în conformitate cu prevederile pct. 63 şi 64 din prezentul Regulament.
1) să verifice dacă, în condiţii normale de utilizare, performanţele dispozitivului sînt în conformitate cu cele prevăzute în pct. 2 din anexa nr. 1 la prezentul Regulament;
2) să determine orice efecte secundare nedorite, în condiţii normale de utilizare, şi să evalueze dacă acestea constituie riscuri acceptabile în raport cu performanţele preconizate ale dispozitivului.
2. Consideraţii etice
Investigaţiile clinice se efectuează în conformitate cu Declaraţia de la Helsinki, aprobată la cea de-a 18-a Adunare Medicală Mondială de la Helsinki, Finlanda, în 1964, modificată la cea de-a 29-a Adunare Medicală Mondială de la Tokio, Japonia, în 1975, şi la cea de-a 35-a Adunare Medicală Mondială de la Veneţia, Italia, în 1983. Este obligatoriu ca toate măsurile cu privire la protecţia subiecţilor umani să fie realizate în spiritul Declaraţiei de la Helsinki. Aceasta include toate etapele investigaţiei clinice, de la prima considerare cu privire la necesitatea şi justificarea studiului pînă la publicarea rezultatelor.
3. Metode:
1) Investigaţiile clinice se efectuează în conformitate cu un plan de investigaţie adecvat, conform nivelului ştiinţific al momentului, astfel definit încît să confirme sau să respingă cele invocate de producător cu privire la dispozitiv; investigaţiile urmează să includă un număr corespunzător de observaţii pentru a garanta validitatea ştiinţifică a concluziilor.
2) Procedurile folosite pentru efectuarea investigaţiilor urmează să fie adecvate dispozitivului destinat examinaării
3) Investigaţiile clinice se efectuează în circumstanţe echivalente condiţiilor normale de utilizare a dispozitivului.
4) Se examinează toate caracteristicile corespunzătoare, inclusiv cele privind siguranţa şi performanţele dispozitivului, precum şi efectele sale asupra pacienţilor.
5) Toate incidentele adverse grave urmează să fie înregistrate complet şi notificate de îndată tuturor autorităţilor competente ale statelor în care are loc investigaţia clinică.
6) Investigaţiile se efectuează sub responsabilitatea unui practician medical calificat în mod corespunzător sau a unei persoane autorizate, într-un mediu adecvat.
Specialistul medical beneficiază de acces la datele tehnice referitoare la dispozitiv.
7) Raportul scris, semnat de către medicul specialist responsabil, cuprinde o evaluare critică a tuturor datelor obţinute în timpul investigaţiei clinice.
2. Organismul recunoscut şi personalul său trebuie să efectueze operaţiunile de evaluare şi verificare la cel mai înalt standard de integritate profesională şi competenţă tehnică şi trebuie să fie în afara oricăror presiuni sau influenţe, în special de natură financiară, care ar putea influenţa decizia acestora sau rezultatele inspecţiei, în special din partea persoanelor ori a grupurilor de persoane interesate de rezultatul verificărilor.
3. Organismul recunoscut urmează să realizeze toate sarcinile prevăzute în anexele nr. 2–5 la prezentul Regulament, pentru care a fost recunoscut, indiferent dacă aceste sarcini sînt executate de organismul respectiv sau sub responsabilitatea sa. În special, trebuie să aibă la dispoziţie personalul necesar şi să posede facilităţile necesare pentru îndeplinirea corespunzătoare a sarcinilor tehnice şi administrative impuse de evaluare şi verificare.
Organismul recunoscut trebuie, de asemenea, să aibă acces la echipamentele necesare pentru verificările cerute.
4. Personalul responsabil de operaţiunile de control deţin:
1) pregătire profesională temeinică, care să acopere toate operaţiile de evaluare şi verificare pentru care a fost recunoscut organismul;
2) cunoştinţe corespunzătoare privind cerinţele operaţiunilor de control pe care le efectuează şi experienţă corespunzătoare în acest domeniu;
3) capacitatea necesară pentru întocmirea certificatelor, înregistrărilor şi rapoartelor, pentru a demonstra că s-au efectuat respectivele operaţiuni de control.
5. Imparţialitatea personalului care realizează inspecţia trebuie să fie garantată. Remuneraţia acestuia nu influenţează şi nu depinde de numărul inspecţiilor efectuate şi nici de rezultatul acestor inspecţii.
6. Organismul recunoscut trebuie să încheie o asigurare de răspundere civilă, cu excepţia cazului în care această răspundere este asigurată de către stat prin lege.
7. Personalul organismului recunoscut este obligat să păstreze secretul profesional cu privire la toate informaţiile obţinute pe durata sarcinilor ce-i revin prin prezentul Regulament sau prin orice prevederi ale legislaţiei în vigoare. Personalul organismului nu are obligaţia păstrării secretului profesional faţă de Agenţie.
2. Pentru produsele care fac obiectul mai multor reglementări tehnice ce prevăd aplicarea mărcii naţionale de conformitate SM, marca respectivă semnifică faptul că produsele în cauză sînt conforme cu prevederile tuturor reglementărilor tehnice aplicabile.
3. Marca naţională de conformitate SM este formată din literele S şi M, care simbolizează „securitate conform cerinţelor esenţiale” şi, respectiv, „Moldova”. Simbolul grafic al mărcii naţionale de conformitate SM este prezentat în figura 1.

4. Dimensiunile mărcii naţionale de conformitate SM trebuie să corespundă întocmai celor specificate în figura 2.
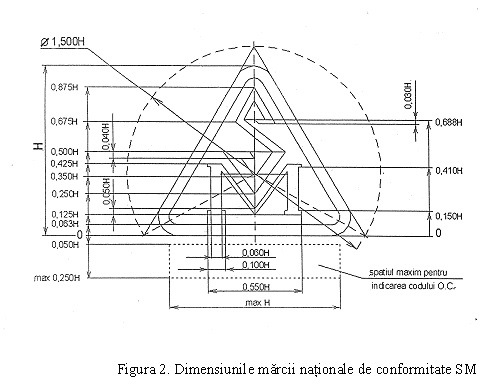
5. În cazul în care marca naţională de conformitate SM urmează a fi mărită sau micşorată, se vor respecta dimensiunile specificate în figura 2.
6. Marca naţională de conformitate SM se execută în alb-negru sau într-o singură culoare, în contrast cu fondul.
7. Pe orice produs din domeniul reglementat va fi aplicată marca naţională de conformitate SM.
8. Marca naţională de conformitate SM se execută prin orice procedeu tehnologic care asigură obţinerea unei imagini clare şi durabile a ei pe toată perioada de utilizare a produselor respective marcate.
9. Marca naţională de conformitate SM este însoţită de numărul de identificare al organismului de evaluare a conformităţii recunoscut, care a fost antrenat în faza de evaluare respectivă, conform prevederilor reglementărilor tehnice aplicabile. Numărul de identificare al organismului de evaluare a conformităţii desemnat se scrie la distanţa de 5% din înălţimea mărcii, sub desenul grafic al acesteia, simetric faţă de axa verticală, cu înălţimea literelor (cifrelor) pînă la 15% din înălţimea mărcii.
10. Marca naţională de conformitate SM şi numărul de identificare al organismului respectiv de evaluare a conformităţii desemnat se aplică de către producător sau de către reprezentantul său.
11. Marca naţională de conformitate SM este utilizată de către producător sau de către reprezentantul său în mod gratuit.
12. Pe un produs, concomitent cu marca naţională de conformitate SM, pot fi aplicate mărci diferite (de exemplu, mărci ce indică conformitatea cu standardele naţionale sau europene sau cu alte reglementări), cu condiţia ca aceste mărci să nu poată fi confundate cu marca naţională de conformitate SM. Aceste mărci pot fi aplicate cu condiţia ca lizibilitatea şi vizibilitatea mărcii naţionale de conformitate SM să nu fie afectate.
13. Dacă organul de supraveghere şi control stabileşte că marca naţională de conformitate SM a fost aplicată neadecvat, producătorul, reprezentantul său sau (în mod excepţional dacă reglementarea tehnică aplicabilă prevede astfel) persoana responsabilă de plasarea produsului respectiv pe piaţă are obligaţia să înlăture neconformităţile respective.
14. În cazul în care neconformitatea nu este înlăturată, organul de supraveghere şi control restricţionează sau interzice distribuirea sub orice formă a produsului respectiv ori asigură retragerea de pe piaţă a acestuia, în conformitate cu prevederile legislaţiei în vigoare.
În temeiul prevederilor art. 54 din Legea ocrotirii sănătăţii nr. 411-XIII din 28 martie 1995 (Monitorul Oficial al Republicii Moldova, 1995, nr. 34, art. 373), cu modificările şi completările ulterioare, art. 4 alin. (4), art. 5, art. 13, art. 21 alin. (3), art. 25 alin. (1) din Legea nr. 92 din 26 aprilie 2012 cu privire la dispozitivele medicale (Monitorul Oficial al Republicii Moldova, 2012, nr. 149-154, art. 480), Guvernul HOTĂRĂŞTE:
1. Se aprobă Regulamentul privind condiţiile de plasare pe piaţă a dispozitivelor medicale implantabile active (se anexează).
2. Controlul asupra executării prezentei hotărîri se pune în sarcina Ministerului Sănătăţii şi Ministerului Mediului.
PRIM-MINISTRU Iurie LEANCĂ
Contrasemnează:
Viceprim-ministru,
ministrul economiei Valeriu Lazăr
Ministrul sănătăţii Andrei Usatîi
Ministrul mediului Gheorghe Şalaru
Nr. 410. Chişinău, 4 iunie 2014.
Aprobat
prin Hotărîrea Guvernului nr. 410
din 4 iunie 2014
prin Hotărîrea Guvernului nr. 410
din 4 iunie 2014
REGULAMENT
privind condiţiile de plasare pe piaţă a dispozitivelor
medicale implantabile active
Regulamentul privind condiţiile de plasare pe piaţă a dispozitivelor medicale implantabile active (în continuare – Regulament) transpune Directiva 90/385/CEE a Consiliului Europei din 20 iunie 1990 privind dispozitivele medicale implantabile active, publicată în Jurnalul Oficial al Comunităţii Europene L 189 din 20 iunie 1990.privind condiţiile de plasare pe piaţă a dispozitivelor
medicale implantabile active
CAPITOLUL I
Dispoziţii generale
1. Prezentul Regulament se aplică dispozitivelor medicale implantabile active.Dispoziţii generale
2. În sensul prezentului Regulament se utilizează terminologia definită în Legea nr. 92 din 26 aprilie 2012 cu privire la dispozitivele medicale, Legea nr. 235 din 1 decembrie 2011 privind activităţile de acreditare şi de evaluare a conformităţii, Legea nr. 1409-XIII din 17 decembrie 1997 cu privire la medicamente, precum şi următoarea noţiune:
organism de evaluare a conformităţii recunoscut – organism de evaluare a conformităţii acreditat, persoană juridică cu sediul în Republica Moldova şi recunoscut de Ministerul Sănătăţii pentru activitatea de evaluare a conformităţii, conform actelor normative cu privire la dispozitivele medicale, aprobate de Guvern.
3. În cazul în care un dispozitiv medical implantabil activ este destinat administrării unui medicament în sensul definiţiei din art. 2 din Legea nr. 92 din 26 aprilie 2012 cu privire la dispozitivele medicale, acesta este reglementat prin prezentul Regulament, fără a aduce atingere definiţiei privitor la medicamente.
4. În cazul în care un dispozitiv medical implantabil activ încorporează, ca parte integrantă, o substanţă care, dacă este utilizată separat, se consideră medicament în sensul art. 3 din Legea nr. 1409-XIII din 17 decembrie 1997 cu privire la medicamente şi care acţionează asupra organismului uman printr-o acţiune auxiliară celei a dispozitivului, acest dispozitiv se evaluează şi se înregistrează conform prevederilor prezentului Regulament.
5. În cazul în care un dispozitiv medical implantabil activ încorporează, ca parte integrantă, o substanţă care, dacă este utilizată separat, se consideră constituent al unui medicament sau un medicament derivat din sînge uman sau din plasmă umană (în continuare – derivat din sînge uman) şi care poate acţiona asupra organismului uman printr-o acţiune auxiliară celei a dispozitivului, acesta este evaluat şi înregistrat în conformitate cu prevederile prezentului Regulament.
6. Prezentul Regulament constituie o reglementare specifică în sensul pct.4 din Reglementarea tehnică „Compatibilitatea electromagnetică a echipamentelor”, aprobată prin Hotărîrea Guvernului nr. 95 din 4 februarie 2008 „Cu privire la aprobarea Reglementării tehnice „Compatibilitatea electromagnetică a echipamentelor”.
7. Prezentul Regulament nu se aplică:
1) medicamentelor;
2) sîngelui uman, produselor din sînge, plasmei sau celulelor sangvine de origine umană ori dispozitivelor care încorporează în momentul introducerii lor pe piaţă astfel de produse din sînge, plasmă sau celule, cu excepţia dispozitivelor prevăzute la pct. 5 din prezentul Regulament;
3) transplanturilor, ţesuturilor sau celulelor de origine umană, precum şi produselor care încorporează sau derivă din ţesuturi ori celule de origine umană, cu excepţia dispozitivelor prevăzute la pct. 5 din prezentul Regulament;
4) transplanturilor, ţesuturilor sau celulelor de origine animală, cu excepţia cazurilor în care un dispozitiv este fabricat prin utilizarea de ţesuturi de origine animală neviabile sau de produse neviabile derivate din ţesuturi de origine animală.
CAPITOLUL II
Introducerea pe piaţă a dispozitivelor medicale
implantabile active
8. Agenţia Medicamentului şi Dispozitivelor Medicale (în continuare – Agenţia) asigură plasarea pe piaţă şi/sau punerea în funcţiune a dispozitivelor implantabile active, numai dacă acestea respectă cerinţele stabilite prin prezentul Regulament, atunci cînd sînt furnizate, implantate şi/sau instalate corespunzător, întreţinute şi utilizate corect, în conformitate cu scopul propus. Introducerea pe piaţă a dispozitivelor medicale
implantabile active
9. Dispozitivele medicale implantabile active (în continuare – dispozitive) la care se face referire în pct. 2-5 din prezentul Regulament asigură conformitatea cu cerinţele esenţiale prevăzute în anexa nr. 1 la prezentul Regulament, care le sînt aplicabile, conform scopului propus al acestora.
10. Se introduc pe piaţă şi se pun în funcţiune numai dispozitivele conforme cu prevederile prezentului Regulament şi care poartă marcajul european de conformitate CE, prevăzut la pct. 42-44 din prezentul Regulament, care demonstrează că aceste dispozitive au fost supuse evaluării conformităţii potrivit capitolului V din prezentul Regulament.
11. Nu pot face obiectul restricţionării:
1) punerea la dispoziţia medicilor calificaţi în mod corespunzător sau a persoanelor autorizate în acest scop a dispozitivelor destinate investigaţiilor clinice, dacă acestea îndeplinesc condiţiile prevăzute în capitolul VI şi în anexa nr. 6 la prezentul Regulament;
2) introducerea pe piaţă şi punerea în funcţiune a dispozitivelor fabricate la comandă, dacă acestea îndeplinesc condiţiile prevăzute în anexa nr. 6 la prezentul Regulament şi sînt însoţite de declaraţia menţionată în această anexă, care se pune la dispoziţia pacientului specific identificat. Dispozitivele menţionate nu poartă marcajul CE de conformitate.
12. La tîrgurile, expoziţiile, demonstraţiile, întrunirile ştiinţifice şi tehnice şi altele asemenea organizate pe teritoriul Republicii Moldova, dispozitivele care nu sînt conforme cu prevederile prezentului Regulament pot fi expuse, cu condiţia să poarte o inscripţionare vizibilă, care să indice în mod clar că nu pot fi comercializate sau puse în funcţiune înainte de a fi aduse la conformitate cu prevederile prezentului Regulament.
13. Dacă un dispozitiv este pus în funcţiune, informaţiile prevăzute la pct. 11 alin. 8) şi 9) şi pct. 12 din anexa nr. 1 la prezentul Regulament urmează să fie furnizate în limba de stat.
14. În cazul în care un dispozitiv face obiectul mai multor reglementări tehnice care prevăd aplicarea marcajului de conformitate, marcajul semnifică faptul că dispozitivul este conform cu prevederile tuturor reglementărilor tehnice respective.
15. Dacă una sau mai multe dintre reglementările tehnice prevăzute la pct. 12 permit producătorului, pentru o perioadă tranzitorie, să aleagă reglementările pe care să le aplice, marcajul de conformitate semnifică faptul că dispozitivele satisfac numai prevederile reglementărilor aplicate de producător.
16. În cazul prevăzut la pct.13 din prezentul Regulament, elementele de identificare ale reglementărilor tehnice aplicate de producător se indică în documentele, notele sau instrucţiunile cerute de reglementările care însoţesc dispozitivul. Documentele, notele sau instrucţiunile care însoţesc dispozitivul trebuie să fie accesibile, fără a fi necesară distrugerea ambalajului care asigură sterilitatea dispozitivului.
17. Se consideră, că cerinţele esenţiale prevăzute în anexa nr. 1 la prezentul Regulament sînt îndeplinite, dacă dispozitivele medicale sînt conforme cu specificaţiile tehnice din standardele naţionale conexe la prezentul Regulament, care adoptă standardele europene armonizate.
În sensul prezentului Regulament, referirea la standardele armonizate include, de asemenea, monografiile Farmacopeii Europene, în special în ceea ce priveşte interacţiunea dintre medicamente şi materialele utilizate în dispozitivele care conţin astfel de medicamente, ale căror referinţe au fost publicate în Monitorul Oficial al Republicii Moldova.
În situaţia în care Agenţia constată că standardele naţionale conexe nu satisfac în totalitate cerinţele esenţiale prevăzute în anexa nr. 1 la prezentul Regulament, aceasta va aplica una din prevederile art. 62 din Legea nr. 590-XIII din 22 septembrie 1995 cu privire la standardizare.
CAPITOLUL III
Clauza de salvgardare
18. În situaţia în care se constată că dispozitivele medicale implantabile active şi cele fabricate la comandă, corect puse în funcţiune şi utilizate corespunzător scopului propus, pot compromite sănătatea şi/sau securitatea pacienţilor, a utilizatorilor ori, după caz, a altor persoane, Agenţia asigură retragerea de pe piaţă, interzicerea acestor dispozitive sau restrîngerea introducerii lor pe piaţă ori punerii lor în funcţiune.Clauza de salvgardare
Producătorul este responsabil pentru activităţile ulterioare retragerii/interzicerii dispozitivelor medicale.
19. Agenţia informează producătorul sau reprezentantul autorizat al acestuia, importatorul/distribuitorul, instituţiile medico-sanitare sau alţi utilizatori cu privire la măsurile prevăzute la pct. 18 din prezentul Regulament, indicînd motivele pentru care a luat această decizie şi, în special, dacă neconformitatea cu prezentul Regulament se datorează următoarelor cauze:
1) neîndeplinirea cerinţelor esenţiale menţionate în anexa nr. 1 la prezentul Regulament, în cazul în care dispozitivul nu îndeplineşte în totalitate sau în parte standardele prevăzute la pct. 17 alineatul unu din prezentul Regulament;
2) aplicarea incorectă a standardelor menţionate la pct. 17 alineatul unu din prezentul Regulament;
3) unele deficienţe ale standardelor.
20. În cazul în care dispozitivele medicale necorespunzătoare poartă marcajul CE, Agenţia are obligaţia de a anunţa Comisia Europeană în termen de 72 de ore de la data constatării.
CAPITOLUL IV
Vigilenţa. Informarea privind incidentele datorate
dispozitivelor introduse pe piaţă
21. Agenţia înregistrează şi evaluează, în mod centralizat, orice informaţie privind următoarele incidente semnalate în legătură cu aplicarea dispozitivelor:Vigilenţa. Informarea privind incidentele datorate
dispozitivelor introduse pe piaţă
1) orice funcţionare defectuoasă sau deteriorare a caracteristicilor şi a performanţelor unui dispozitiv, precum şi orice etichetare sau instrucţiuni de utilizare inadecvate, care pot să conducă ori au condus la decesul sau deteriorarea severă a stării de sănătate a unui pacient ori utilizator;
2) orice cauză de ordin tehnic sau medical vizînd caracteristicile ori performanţele unui dispozitiv, care, din motivele prevăzute la subpct. 1) din prezentul punct, conduce la retragerea sistematică de pe piaţă de către producător a dispozitivelor de acelaşi tip.
22. Obligaţia de a anunţa Agenţia despre incidentele menţionate la pct. 21 revine producătorului sau reprezentantului său autorizat, importatorului, distribuitorului, personalului medical, instituţiilor medico-sanitare sau altor utilizatori.
23. În cazul în care informarea despre incidentele menţionate la pct. 21 a fost transmisă de personalul medical, instituţiile medico-sanitare sau de către alţi utilizatori, Agenţia informează producătorul dispozitivului în cauză ori reprezentantul său autorizat cu privire la incident.
24. După efectuarea unei evaluări, dacă este posibil, împreună cu producătorul sau cu reprezentantul său autorizat, respectînd prevederile capitolului III din prezentul Regulament, Agenţia informează imediat autorităţile competente din alte state cu care are încheiate acorduri referitor la măsurile care au fost luate sau care sînt avute în vedere pentru minimizarea riscului de reproducere a incidentelor menţionate la pct. 21, inclusiv informaţii referitoare la incidentele depistate.
CAPITOLUL V
Evaluarea conformităţii
25. În cazul dispozitivelor, altele decît dispozitivele fabricate la comandă sau cele destinate investigaţiilor clinice, producătorul, în scopul aplicării marcajului de conformitate, urmează una dintre următoarele proceduri:Evaluarea conformităţii
1) procedura referitoare la declaraţia de conformitate, prevăzută în anexa nr. 2 la prezentul Regulament;
2) procedura referitoare la examinarea CE de tip, prevăzută în anexa nr. 3 la prezentul Regulament, asociată cu:
a) procedura referitoare la verificarea unităţii de produs, prevăzută în anexa nr. 4 la prezentul Regulament;
b) procedura referitoare la declaraţia de conformitate, prevăzută în anexa nr. 5 la prezentul Regulament.
26. În cazul dispozitivelor fabricate la comandă, producătorul trebuie să emită declaraţia privind dispozitivele cu scopuri speciale, conform anexei nr. 6 la prezentul Regulament, înainte de introducerea pe piaţă a fiecărui dispozitiv.
27. În cazuri argumentate, procedurile prevăzute în anexele nr. 3, 4 şi 6 la prezentul Regulament se aplică de reprezentantul autorizat al producătorului stabilit în Republica Moldova.
28. Înregistrările şi corespondenţa referitoare la procedurile menţionate la pct. 25-27 din prezentul Regulament se redactează în limba de stat.
29. Evaluarea conformităţii dispozitivelor medicale se efectuează de organisme de evaluare a conformităţii, acreditate în condiţiile Legii nr. 235 din 1 decembrie 2011 privind activităţile de acreditare şi de evaluare a conformităţii şi recunoscute de Ministerul Sănătăţii, în baza criteriilor stabilite de prezentul Regulament.
În cursul procedurii de evaluare a conformităţii pentru un dispozitiv, producătorul şi/sau organismul de evaluare a conformităţii recunoscut (în continuare – organism recunoscut) ţine cont de rezultatele obţinute în urma oricăror operaţiuni de evaluare şi verificare care au fost efectuate potrivit prevederilor prezentului Regulament într-o fază intermediară de fabricaţie.
30. În cazul în care procedura de evaluare a conformităţii implică intervenţia unui organism recunoscut, producătorul sau reprezentantul său autorizat stabilit în Republica Moldova este în drept să se adreseze unui organism la alegere, corespunzător sarcinilor în legătură cu care acesta a fost recunoscut.
31. Organismul recunoscut solicită orice informaţii sau date suplimentare care sînt necesare pentru a stabili şi a menţine atestarea conformităţii în funcţie de procedura aleasă.
32. Deciziile adoptate de organismul recunoscut potrivit prevederilor anexelor nr. 2, 3 şi 5 la prezentul Regulament au o valabilitate maximă de 5 ani şi urmează a fi prelungite, pentru perioade suplimentare de cel mult 5 ani, la cererea înaintată de către producătorul sau reprezentantul său autorizat, la o dată stabilită în contractul semnat de ambele părţi.
33. Agenţia autorizează, în cazuri deosebite (cataclisme, catastrofe, epidemii, epizootii, intoxicaţii în masă, în alte cazuri ce ameninţă sănătatea oamenilor; absenţa pe piaţă a analogurilor sau a substituenţilor dispozitivelor medicale), punerea la dispoziţie pe piaţă şi punerea în funcţiune a dispozitivelor neautorizate în Republica Moldova, dar autorizate în ţara de origine, pentru care nu au fost efectuate procedurile prevăzute la pct. 27-31 din prezentul Regulament şi a căror utilizare este în interesul protecţiei sănătăţii.
După expirarea perioadei de forţă majoră, dar nu mai mult de 1 an de la declanşarea acesteia, dispozitivele sus-menţionate urmează să fie supuse în mod obligatoriu procedurii de autorizare în Republica Moldova. În caz contrar, Agenţia îşi rezervă dreptul de a dispune retragerea de pe piaţă şi de a interzice plasarea pe piaţă a dispozitivelor respective.
34. Agenţia asigură realizarea acţiunilor în următoarele situaţii:
1) atunci cînd constată că stabilirea conformităţii unui dispozitiv sau a unei grupe de dispozitive se efectuează prin excepţie de la prevederile capitolului V din prezentul Regulament, prin aplicarea exclusivă a unei proceduri respective, selectată dintre cele prevăzute la capitolul V din prezentul Regulament;
2) atunci cînd constată că este necesară o decizie pentru a determina dacă un anumit produs sau grup de produse se încadrează în definiţia prevăzută în Legea nr. 92 din 26 aprilie 2012 cu privire la dispozitivele medicale: „dispozitiv medical”, „dispozitiv medical implantabil activ”, „dispozitiv fabricat la comandă” şi „dispozitiv destinat investigaţiei clinice”.
CAPITOLUL VI
Investigaţia clinică
35. În cazul dispozitivelor destinate investigaţiilor clinice, producătorul sau reprezentantul autorizat stabilit în Republica Moldova prezintă Agenţiei, cu cel puţin 60 de zile înainte de începerea investigaţiilor, declaraţia prevăzută în anexa nr. 6 la prezentul Regulament.Investigaţia clinică
36. Producătorul este în drept să înceapă investigaţiile clinice la sfîrşitul perioadei de 60 de zile de la notificare, doar dacă pînă la sfîrşitul acestei perioade Agenţia îi comunică acestuia decizia de acceptare.
Agenţia va autoriza producătorii pentru începerea investigaţiilor clinice respective înaintea expirării perioadei de 60 de zile, dacă Comisia pentru dispozitive medicale a emis un aviz favorabil privind programul investigaţiei în cauză, cuprinzînd şi analiza sa referitoare la planul investigaţiei clinice.
37. Agenţia are obligaţia să asigure sănătatea publică şi realizarea reglementărilor în domeniu:
1) în cazul în care investigaţia clinică este refuzată sau suspendată de Agenţie, ultima comunică solicitantului decizia sa şi motivele care au stat la baza acesteia;
2) în cazul în care Agenţia a solicitat o modificare semnificativă sau întreruperea temporară a unei investigaţii clinice, aceasta informează solicitantul cu privire la acţiunile sale şi motivele pentru acţiunile întreprinse.
38. Producătorul sau reprezentantul său autorizat notifică Agenţia cu privire la sfîrşitul investigaţiei clinice, cu o justificare în cazul unei încetări anticipate.
În cazul unei încetări anticipate a investigaţiei clinice din motive de securitate, Agenţia comunică solicitantului această notificare.
Producătorul sau reprezentantul său autorizat pune la dispoziţia Agenţiei raportul prevăzut în secţiunea a II-a, pct.3, subpct. 7) din anexa nr. 7 la prezentul Regulament.
39. Investigaţiile clinice se desfăşoară potrivit prevederilor anexei nr. 7 la prezentul Regulament.
CAPITOLUL VII
Înregistrarea dispozitivelor
40. Producătorii cu sediul în Republica Moldova, care introduc pe piaţă dispozitive în nume propriu, conform procedurii prevăzute la pct. 26 din prezentul Regulament, au obligaţia de a se înregistra la Agenţie, furnizînd date cu privire la adresa sediului juridic şi la descrierea dispozitivelor care fac obiectul activităţii acestora, în scopul introducerii informaţiei în baza de date privind dispozitivele medicale a Agenţiei.Înregistrarea dispozitivelor
41. Pentru toate dispozitivele, Agenţia se informează cu privire la toate datele care permit identificarea acestor dispozitive, împreună cu eticheta şi instrucţiunile de utilizare, atunci cînd dispozitivele sînt puse în funcţiune pe teritoriul Republicii Moldova.
42. În cazul în care un producător care introduce pe piaţă un dispozitiv în nume propriu nu are sediu juridic în Republica Moldova, acesta desemnează un reprezentant autorizat în Republica Moldova.
43. În cazul dispozitivelor prevăzute la pct. 40 din prezentul Regulament, reprezentantul autorizat cu sediu juridic în Republica Moldova informează Agenţia cu privire la toate datele menţionate la pct. 40 din prezentul Regulament.
44. Agenţia informează, la cerere, autorităţile competente din alte state cu care are încheiate acorduri cu privire la datele prevăzute la pct. 40 din prezentul Regulament, furnizate de către producător sau de către reprezentantul său autorizat.
45. Datele înregistrate, potrivit prevederilor prezentului Regulament, se stochează în baza de date privind dispozitivele medicale a Agenţiei.
46. Baza de date privind dispozitivele medicale a Agenţiei cuprinde informaţii referitoare la:
1) înregistrarea producătorilor şi dispozitivelor, potrivit prevederilor pct. 40;
2) certificatele emise, modificate, suplimentate, suspendate, retrase sau respinse potrivit prevederilor din anexele nr. 2-5 la prezentul Regulament;
3) procedura de vigilenţă prevăzută în capitolul IV la prezentul Regulament;
4) investigaţiile clinice prevăzute în capitolul VI din prezentul Regulament.
Datele prevăzute la prezentul punct se furnizează în format standard.
47. Normele de procedură pentru aplicarea prevederilor pct. 45-46 din prezentul Regulament se aprobă prin ordin al Ministerului Sănătăţii.
CAPITOLUL VIII
Măsurile speciale de monitorizare a sănătăţii
48. Agenţia dispune, potrivit capitolului III din prezentul Regulament, retragerea, limitarea sau interzicerea unui anumit dispozitiv ori grup de dispozitive de pe piaţă, sau impunerea asupra acestora a unor cerinţe specifice pentru introducerea lor pe piaţă ori punerea lor în funcţiune, în scopul protecţiei sănătăţii şi/sau pentru a asigura respectarea cerinţelor de sănătate publică.Măsurile speciale de monitorizare a sănătăţii
49. Agenţia informează despre măsurile aplicate potrivit pct. 48 Ministerul Sănătăţii, producătorul şi organismul de evaluare a conformităţii recunoscut.
CAPITOLUL IX
Organismele de evaluare a conformităţii recunoscute
50. Pentru evaluarea conformităţii dispozitivelor medicale, conform procedurilor de evaluare a conformităţii menţionate în capitolul V, Agenţia, aplicînd criteriile minime obligatorii prevăzute în anexa nr. 8 la prezentul Regulament, propune Ministerului Sănătăţii spre recunoaştere organismele de evaluare a conformităţii, acreditate de Centrul Naţional de Acreditare.Organismele de evaluare a conformităţii recunoscute
În cazul în care Agenţia constată că un organism recunoscut nu corespunde criteriilor specificate, care au stat la baza recunoaşterii, propune Ministerului Sănătăţii anularea ordinului de recunoaştere şi informează Centrul Naţional de Acreditare.
51. Organismul recunoscut şi producătorul sau reprezentantul său autorizat stabilesc de comun acord termenele-limită pentru finalizarea activităţilor de evaluare şi verificare prevăzute în anexele nr. 2-5 la prezentul Regulament.
52. Organismul recunoscut informează Agenţia referitor la toate certificatele emise, modificate, suplimentate, suspendate, retrase sau refuzate, precum şi celelalte organisme recunoscute în sensul prezentului Regulament cu privire la certificatele retrase, suspendate sau refuzate şi, la cerere, cu privire la certificatele eliberate. Organismul recunoscut oferă, de asemenea, la cererea Agenţiei, toate informaţiile suplimentare relevante.
53. În cazul în care un organism recunoscut constată că cerinţele relevante din prezentul Regulament nu au fost îndeplinite sau au încetat să mai fie îndeplinite de producător sau că un certificat nu ar fi trebuit să fie emis, atunci, bazîndu-se pe principiul proporţionalităţii, suspendă sau retrage certificatul emis ori impune restricţii asupra acestuia pînă cînd conformitatea cu aceste cerinţe este asigurată de către producător.
În cazul suspendării sau retragerii certificatului ori al impunerii de restricţii sau în cazurile în care este necesară o intervenţie din partea autorităţii competente, organismul recunoscut informează Agenţia cu privire la acest fapt.
Agenţia informează părţile interesate cu privire la măsurile luate, conform alineatului unu din prezentul punct.
54. Organismul recunoscut oferă, la cerere, orice informaţii sau documente relevante, inclusiv documentele bugetare, necesare pentru a-i permite Agenţiei să verifice respectarea criteriilor menţionate în anexa nr. 8 la prezentul Regulament.
CAPITOLUL X
Marcajul de conformitate
55. Dispozitivele, cu excepţia celor fabricate la comandă şi a celor destinate investigaţiilor clinice, considerate că satisfac cerinţele esenţiale prevăzute la pct. 9 din prezentul Regulament, poartă în momentul introducerii pe piaţă marcajul de conformitate.Marcajul de conformitate
56. Marcajul de conformitate, potrivit prevederilor anexei nr. 9 la prezentul Regulament, se aplică vizibil, lizibil şi de neşters pe ambalajul steril şi, dacă este cazul, pe ambalajul comercial şi pe instrucţiunile de utilizare.
Marcajul de conformitate este însoţit de numărul de identificare al organismului recunoscut, care poartă răspunderea pentru aplicarea procedurilor prevăzute în anexele nr. 2, 4 şi 5 la prezentul Regulament.
57. Este interzisă aplicarea de marcaje care poate induce în eroare părţile terţe cu privire la înţelesul sau forma grafică a marcajului de conformitate.
Se aplică, de asemenea, orice alt marcaj pe ambalaj sau pe instrucţiunile care însoţesc dispozitivul, cu condiţia ca acesta să nu afecteze vizibilitatea şi claritatea marcajului de conformitate.
58. Fără a aduce atingere prevederilor capitolului III din prezentul Regulament, în cazul în care Agenţia stabileşte că marcajul de conformitate a fost aplicat în mod necorespunzător sau lipseşte, încălcînd prezentul Regulament, producătorul sau reprezentantul său autorizat este obligat să lichideze încălcările depistate.
Dacă se menţine situaţia de neconformitate prevăzută la alineatul unu din prezentul punct, Agenţia limitează sau interzice introducerea pe piaţă a produsului în cauză ori se asigură că este retras de pe piaţă, în conformitate cu procedura prevăzută la capitolul III din prezentul Regulament.
59. Prevederile pct. 58 din prezentul Regulament se aplică şi atunci cînd marcajul de conformitate a fost aplicat produselor care nu fac obiectul prezentului Regulament, dar în conformitate cu prevederile prezentului Regulament.
CAPITOLUL XI
Deciziile de respingere sau de restrîngere
şi obligaţia de confidenţialitate
Secţiunea 1
Deciziile de respingere sau de restrîngere
60. Pentru orice decizie adoptată conform prezentului Regulament, prin care se respinge sau se restrînge introducerea pe piaţă, punerea în funcţiune a unui dispozitiv, efectuarea unei investigaţii clinice sau prin care se retrag dispozitive de pe piaţă, se expun motivele care stau la baza acesteia.Deciziile de respingere sau de restrîngere
şi obligaţia de confidenţialitate
Secţiunea 1
Deciziile de respingere sau de restrîngere
61. Deciziile prevăzute la pct. 60 din prezentul Regulament sînt aduse la cunoştinţă fără întîrziere producătorului sau reprezentantului său autorizat, care va fi informat totodată cu privire la căile de atac pe care le are la dispoziţie, conform reglementărilor în vigoare, cît şi cu privire la termenul-limită pînă la care pot fi exercitate căile de atac.
62. În cazul unei decizii de natura celor prevăzute la pct. 60, părţile interesate sus-menţionate au posibilitatea de a-şi expune în prealabil punctul de vedere, cu excepţia cazului în care consultarea directă nu este posibilă, datorită urgenţei măsurilor ce urmează să fie adoptate.
Secţiunea a 2-a
Confidenţialitatea
63. Toate părţile implicate în aplicarea prezentului Regulament sînt obligate să respecte caracterul confidenţial al tuturor informaţiilor obţinute în îndeplinirea atribuţiilor lor, cu respectarea legislaţiei în vigoare şi a practicii naţionale cu privire la secretul actului medical.Confidenţialitatea
Prevederile alineatului unu din prezentul punct referitor la asigurarea confidenţialităţii nu aduc atingere obligaţiilor Agenţiei şi ale organismelor recunoscute în ceea ce priveşte informarea reciprocă şi difuzarea avertismentelor şi nici obligaţiilor persoanelor care trebuie să furnizeze informaţii ce cad sub incidenţa legislaţiei penale.
64. Nu sînt considerate confidenţiale următoarele informaţii:
1) informaţiile privind înregistrarea persoanelor responsabile de introducerea pe piaţă a dispozitivelor, conform prevederilor pct. 40 din prezentul Regulament;
2) informaţiile destinate utilizatorilor transmise de producător, reprezentantul autorizat sau distribuitor în legătură cu o anumită măsură, potrivit prevederilor pct. 13 din prezentul Regulament;
3) informaţiile din certificatele emise, modificate, suplimentate, suspendate sau retrase.
CAPITOLUL XII
Supravegherea pieţei
65. Agenţia verifică respectarea prevederilor prezentului Regulament şi este responsabilă pentru supravegherea pieţei dispozitivelor medicale în conformitate cu prezentul Regulament.Supravegherea pieţei
66. Agenţia asigură cooperarea cu autorităţile competente din alte state privind domeniul dispozitivelor medicale implantabile active.
Anexa nr. 1
la Regulamentul privind condiţiile
de plasare pe piaţă a dispozitivelor
medicale implantabile active
la Regulamentul privind condiţiile
de plasare pe piaţă a dispozitivelor
medicale implantabile active
CERINŢE ESENŢIALE
I. Cerinţe generale
1. Dispozitivele sînt proiectate şi fabricate astfel încît, atunci cînd sînt implantate conform condiţiilor şi scopului propus, utilizarea lor să nu compromită starea clinică sau securitatea pacienţilor. Acestea asigură securitatea persoanelor care le implantează sau, după caz, a altor persoane.I. Cerinţe generale
2. Dispozitivele asigură atingerea performanţelor prevăzute de către producător să fie proiectate şi, respectiv, fabricate astfel încît să îndeplinească una sau mai multe dintre funcţiile menţionate la pct. 2 din prezentul Regulament, conform celor specificate de producător.
3. Caracteristicile şi performanţele specificate la pct. 1 şi 2 din prezenta anexă nu trebuie să se deprecieze astfel încît să compromită starea clinică şi securitatea pacienţilor sau, după caz, a altor persoane pe întreaga durată de viaţă a dispozitivului estimată de producător, în condiţiile în care dispozitivul este supus unor solicitări posibile în condiţii normale de utilizare.
4. Dispozitivele urmează să fie proiectate, fabricate şi ambalate astfel încît caracteristicile şi performanţele acestora să nu fie afectate negativ ca urmare a condiţiilor de depozitare şi transport stabilite de producător (temperatură, umiditate etc.).
5. Orice efecte secundare sau stări nedorite trebuie să constituie riscuri acceptabile în raport cu performanţele pentru care este destinat dispozitivul.
6. Demonstrarea conformităţii cu cerinţele esenţiale trebuie să includă o evaluare clinică potrivit prevederilor prevăzute în anexa nr. 7 la prezentul Regulament.
II. Cerinţele de proiectare şi construcţie
7. Soluţiile adoptate de producător pentru proiectarea şi fabricarea dispozitivelor trebuie să corespundă principiilor de securitate, ţinînd seama de nivelul tehnologic general recunoscut la momentul respectiv.8. Dispozitivele implantabile sînt proiectate, fabricate şi ambalate în ambalaje de unică folosinţă, conform procedurilor corespunzătoare, pentru a se asigura că acestea sînt sterile cînd sînt introduse pe piaţă, pe durata depozitării şi a transportului în condiţiile specificate de producător şi că rămîn sterile pînă cînd sînt dezambalate şi implantate.
9. Dispozitivele urmează să fie proiectate şi fabricate astfel încît să se elimine sau să se reducă cît mai mult posibil:
1) riscul de leziuni fizice, în legătură cu caracteristicile lor fizice, inclusiv cele dimensionale;
2) riscurile legate de utilizarea surselor de alimentare cu energie, îndeosebi atunci cînd se foloseşte electricitatea, cu referire, în mod particular, la izolaţie, curenţi de scurgere şi supraîncălzirea dispozitivelor;
3) riscurile legate de condiţiile de mediu previzibile în mod rezonabil, cum sînt cîmpurile magnetice, perturbaţiile electrice externe, descărcările electrostatice, presiunea sau variaţiile de presiune şi acceleraţie;
4) riscurile legate de tratamentele medicale, în particular cele care rezultă din utilizarea defibrilatoarelor sau a echipamentului electrochirurgical de înaltă frecvenţă;
5) riscurile legate de radiaţiile ionizante din substanţele radioactive incluse în dispozitiv;
6) riscurile care pot să apară în condiţiile în care nu sînt posibile întreţinerea şi calibrarea dispozitivelor, inclusiv:
a) creşterea excesivă a curenţilor de scurgere;
b) îmbătrînirea materialelor utilizate;
c) căldura excesivă generată de dispozitiv;
d) scăderea preciziei oricăror mecanisme de măsurare şi control.
10. Dispozitivele urmează să fie proiectate şi fabricate astfel încît să se garanteze caracteristicile şi performanţele cuprinse în capitolul I al prezentei anexe, cu o atenţie specială acordată pentru:
1) alegerea materialelor folosite, mai ales în ceea ce priveşte aspectele legate de toxicitate;
2) compatibilitatea mutuală dintre materialele utilizate şi ţesuturile biologice, celulele şi fluidele organismului, în concordanţă cu utilizarea preconizată a dispozitivului;
3) compatibilitatea dispozitivelor cu substanţele pe care sînt destinate să le administreze;
4) calitatea conexiunilor, în particular cu privire la securitate;
5) fiabilitatea sursei de energie;
6) protecţia împotriva scurgerilor, dacă este cazul;
7) funcţionarea corectă a sistemelor de programare şi control, inclusiv software-ul. În cazul dispozitivelor care încorporează software sau care sînt ele însele software medical, software-ul trebuie validat în conformitate cu nivelul tehnicii la momentul respectiv, luîndu-se în considerare principiile dezvoltării ciclului de viaţă, gestionării riscurilor, validării şi verificării.
11. În cazul în care un dispozitiv încorporează, ca parte integrantă, o substanţă care, dacă este folosită separat, se consideră ca fiind un medicament în sensul definiţiei prevăzute în legislaţia cu privire la medicamente şi care acţionează asupra organismului uman printr-o acţiune auxiliară celei a dispozitivului, calitatea, siguranţa şi utilitatea acelei substanţe se verifică prin analogie cu metodele specificate în normele şi protocoalele analitice, farmacotoxicologice şi clinice referitoare la testarea medicamentelor.
12. În cazul substanţelor menţionate la pct. 11 din prezenta anexă, organismul recunoscut, după ce a verificat utilitatea substanţei ca parte a dispozitivului medical şi ţinînd cont de scopul propus al dispozitivului, solicită avizul ştiinţific al Agenţiei sau al Agenţiei Europene pentru Medicamente (European Medicines Agency, în continuare – EMA), care hotărăşte cu privire la calitatea şi siguranţa substanţei, inclusiv raportul stabilit între beneficiile şi riscurile clinice ale încorporării substanţei în dispozitiv. La emiterea avizului, Agenţia sau EMA ia în considerare procesul de fabricaţie şi datele referitoare la utilitatea încorporării substanţei în dispozitiv, determinate de către organismul recunoscut.
13. În cazul în care un dispozitiv încorporează, ca parte integrantă, un derivat din sînge uman, organismul recunoscut, după ce a verificat utilitatea substanţei ca parte a dispozitivului medical şi ţinînd cont de scopul propus al dispozitivului, solicită avizul ştiinţific al Agenţiei sau al EMA, care hotărăşte cu privire la calitatea şi siguranţa substanţei, inclusiv raportul stabilit între beneficiile şi riscurile clinice ale încorporării derivatului din sînge uman în dispozitiv. La emiterea avizului, Agenţia sau EMA ia în considerare procesul de fabricaţie şi datele referitoare la utilitatea încorporării substanţei în dispozitiv, determinate de către organismul recunoscut.
14. În cazul în care se aduc modificări unei substanţe auxiliare încorporate într-un dispozitiv, în special dacă sînt legate de procesul de fabricaţie a acesteia, organismul recunoscut este informat cu privire la modificări şi consultă agenţia implicată în consultarea iniţială, pentru a confirma menţinerea gradului iniţial de calitate şi siguranţă al substanţei auxiliare. Agenţia ţine seama de datele referitoare la utilitatea încorporării substanţei în dispozitiv, determinate de organismul recunoscut, pentru a se asigura că modificările nu au un impact negativ asupra raportului stabilit între beneficiile şi riscurile adăugării substanţei în dispozitiv.
15. În cazul în care Agenţia, implicată în consultarea iniţială, a obţinut informaţii cu privire la substanţa auxiliară care ar putea avea impact asupra raportului stabilit între beneficiile şi riscurile adăugării substanţei în dispozitiv, aceasta consiliază organismului recunoscut, indiferent dacă informaţiile au sau nu impact asupra raportului stabilit între beneficiile şi riscurile adăugării substanţei în dispozitiv. Organismul recunoscut ţine seama de avizul ştiinţific actualizat şi reanalizează evaluarea sa din cadrul procedurii de evaluare a conformităţii.
16. Dispozitivele şi, după caz, părţile lor componente urmează a fi identificate pentru a permite luarea oricărei măsuri specificate în pct. 20 din prezenta anexă în caz de identificare a unui risc potenţial determinat de dispozitive sau de părţile lor componente.
17. Dispozitivele poartă un cod prin care acestea şi producătorul lor să poată fi identificate fără echivoc (în particular, cu privire la tipul dispozitivului şi anul fabricaţiei); acest cod asigură accesibilitatea pentru a fi citit, dacă este necesar, fără a fi nevoie de o intervenţie chirurgicală.
18. Dacă un dispozitiv sau accesoriile sale poartă instrucţiuni necesare pentru funcţionarea acestuia ori indică parametrii de funcţionare sau de reglare cu ajutorul unui sistem de vizualizare, atunci aceste informaţii asigură claritate pentru utilizator şi, dacă este cazul, pentru pacient.
19. Fiecare dispozitiv este însoţit de următoarele informaţii specifice, scrise lizibil şi care nu pot fi şterse, dacă este cazul, sub forma unor simboluri general recunoscute:
1) pe ambalajul steril:
a) metoda de sterilizare;
b) indicaţia care permite ca ambalajul să fie identificat ca atare;
c) numele şi adresa producătorului;
d) descrierea dispozitivului;
e) cuvintele „exclusiv pentru investigaţie clinică”, dacă dispozitivul este destinat investigaţiilor clinice;
f) cuvintele „dispozitiv fabricat la comandă”, dacă dispozitivul este fabricat la comandă;
g) declaraţia că dispozitivul implantabil este steril;
h) luna şi anul fabricaţiei;
i) indicaţia cu termenul pînă la care este posibilă implantarea dispozitivului în condiţii de securitate;
2) pe ambalajul de vînzare:
a) numele şi adresa producătorului şi a reprezentantului autorizat, în cazul în care producătorul nu are sediul social înregistrat în Republica Moldova;
b) descrierea dispozitivului;
c) scopul propus al dispozitivului;
d) caracteristicile relevante pentru utilizarea acestuia;
e) cuvintele „exclusiv pentru investigaţie clinică”, dacă dispozitivul este destinat investigaţiilor clinice;
f) cuvintele „dispozitiv fabricat la comandă”, dacă dispozitivul este fabricat la comandă;
g) declaraţia că dispozitivul implantabil este steril;
h) luna şi anul fabricaţiei;
i) indicaţia privind termenul pînă la care dispozitivul poate fi implantat în condiţii de securitate;
j) condiţiile de transport şi depozitare pentru dispozitiv;
k) în cazul unui dispozitiv prevăzut la pct. 5 din prezentul Regulament, indicaţia precum că dispozitivul conţine un derivat din sînge uman.
20. La introducerea pe piaţă, fiecare dispozitiv urmează a fi însoţit de:
1) instrucţiunile de utilizare în care să se indice următoarele date specifice:
a) anul autorizării pentru aplicarea marcajului de conformitate;
b) detaliile prevăzute la pct. 19 subpct. 1) şi 2), cu excepţia celor de la lit. h) şi i);
c) performanţele la care se face referire la pct. 2 din prezenta anexă, precum şi orice efecte secundare nedorite;
d) informaţiile care permit medicului să selecteze un dispozitiv adecvat şi software-ul corespunzător, precum şi accesoriile;
e) informaţiile care constituie instrucţiunile de utilizare şi care permit medicului şi, după caz, pacientului să utilizeze corect dispozitivul, software-ul şi accesoriile acestuia, precum şi informaţiile despre natura, domeniul şi periodicitatea verificărilor şi testelor de funcţionare şi, după caz, măsurile de întreţinere;
f) informaţiile care să permită, dacă este cazul, evitarea anumitor riscuri legate de implantarea dispozitivului;
g) informaţiile referitoare la riscurile unor interferenţe reciproce, legate de prezenţa dispozitivului în timpul unor investigaţii sau tratamente specifice; riscurile de interferenţe reciproce reprezintă efecte adverse asupra dispozitivului cauzate de instrumentele prezente la momentul investigaţiilor sau tratamentelor şi viceversa;
h) instrucţiunile necesare în eventualitatea deteriorării ambalajului steril şi, după caz, detaliile privind metodele adecvate de resterilizare;
i) indicaţia, unde este cazul, cu referire la posibilitatea reutilizării dispozitivului, cu condiţia că este recondiţionat sub responsabilitatea producătorului, pentru a corespunde cerinţelor esenţiale;
2) broşura cu instrucţiuni de utilizare trebuie să includă şi detalii care să permită medicului să instruiască pacientul cu privire la contraindicaţiile şi precauţiile necesare. Aceste detalii cuprind în special:
a) informaţiile care să permită stabilirea duratei de viaţă a sursei de energie;
b) precauţiile necesare în cazul în care apar modificări ale performanţelor dispozitivului;
c) precauţiile necesare privind expunerea, în condiţii de mediu previzibile în mod rezonabil, la cîmpuri magnetice, influenţe electrice exterioare, descărcări electrostatice, presiune sau variaţii de presiune, acceleraţie etc.;
d) informaţiile adecvate privind medicamentele pe care dispozitivul este destinat a le administra;
e) data emiterii sau a ultimei revizuiri a instrucţiunilor de utilizare.
21. Confirmarea că dispozitivul îndeplineşte cerinţele cu privire la caracteristicile şi performanţele prevăzute în capitolul I al prezentei anexe, în condiţii normale de funcţionare, şi că evaluarea efectelor secundare sau nedorite se realizează în baza datelor clinice, stabilite conform prevederilor din anexa nr. 7 la prezentul Regulament.
Anexa nr. 2
la Regulamentul privind condiţiile
de plasare pe piaţă a dispozitivelor
medicale implantabile active
la Regulamentul privind condiţiile
de plasare pe piaţă a dispozitivelor
medicale implantabile active
DECLARAŢIA DE CONFORMITATE
privind sistemul integral de asigurare a calităţii
1. Producătorul aplică sistemul calităţii aprobat pentru proiectarea, fabricarea şi inspecţia finală a produselor în cauză, aşa cum este specificat la pct. 3 şi 4 din prezenta anexă, şi este subiectul supravegherii conform pct. 5 din prezenta anexă.privind sistemul integral de asigurare a calităţii
2. Declaraţia de conformitate este procedura prin intermediul căreia producătorul care îndeplineşte obligaţiile impuse la pct. 1 din prezenta anexă asigură şi declară că produsele în cauză îndeplinesc prevederile prezentului Regulament, care le sînt aplicabile.
3. Producătorul sau reprezentantul său autorizat cu sediul în Republica Moldova aplică marcajul de conformitate, potrivit prevederilor capitolului X din prezentul Regulament, şi întocmeşte, în scris, o declaraţie de conformitate. Această declaraţie se referă la unul sau mai multe dispozitive identificate clar prin intermediul denumirii sau al codului produsului ori al altei referinţe lipsite de ambiguitate şi se păstrează de către producător. Marcajul de conformitate este însoţit de numărul de identificare al organismului recunoscut responsabil.
Secţiunea I. Sistemul calităţii
4. Producătorul înaintează o cerere pentru evaluarea sistemului calităţii la un organism recunoscut.Cererea include:
a) toate informaţiile adecvate referitoare la categoria de produse a căror fabricaţie este vizată;
b) documentaţia referitoare la sistemul calităţii;
c) angajamentul producătorului de a îndeplini obligaţiile ce decurg din sistemul calităţii aprobat;
d) angajamentul producătorului de a menţine sistemul calităţii aprobat în aşa fel încît acesta să rămînă adecvat şi eficace;
e) angajamentul producătorului de a institui şi de a menţine actualizat un sistem de supraveghere postvînzare, care să includă dispoziţiile menţionate în anexa nr. 7 la prezentul Regulament. Acest angajament include obligaţia asumată de către producător de a comunica autorităţilor competente următoarele incidente imediat ce a luat cunoştinţă despre acestea:
(i) orice deteriorare a caracteristicilor sau performanţelor dispozitivului;
(ii) orice inexactitate din instrucţiunile de utilizare care ar putea conduce ori au condus la decesul unui pacient sau la deteriorarea stării de sănătate a acestuia;
(iii) orice argument tehnic sau medical care are ca rezultat retragerea de pe piaţă a unui dispozitiv de către producător.
5. Aplicarea sistemului calităţii asigură conformitatea dispozitivelor cu prevederile prezentului Regulament, care le sînt aplicabile, în toate etapele, de la proiectare pînă la controalele finale.
6. Toate elementele, cerinţele şi dispoziţiile adoptate de producător pentru sistemul calităţii sînt documentate în mod sistematic şi ordonat, sub formă de proceduri şi declaraţii scrise privind politica de calitate. Această documentaţie a sistemului calităţii asigură posibilitatea interpretării uniforme a politicilor calităţii şi a procedurilor, cum ar fi: programe ale calităţii, planuri ale calităţii, manuale ale calităţii şi înregistrări ale calităţii. Aceasta include, în special, documentele, datele şi înregistrările corespunzătoare generate de procedurile menţionate la lit.c) din prezentul punct.
Documentaţia include, în special, o descriere adecvată a:
a) obiectivelor producătorului privind calitatea;
b) organizării afacerilor şi, în special:
(i) a structurilor organizatorice, a responsabilităţilor personalului de conducere şi a autorităţii acesteia în legătură cu organizarea, în cazul în care este vizată calitatea proiectării şi fabricaţia produselor;
(ii) a metodelor de monitorizare a funcţionării eficiente a sistemului calităţii şi, în special, capacitatea acestuia de a determina calitatea dorită a proiectului şi a produselor, inclusiv controlul produselor care nu sînt conforme;
(iii) în cazul în care proiectarea, fabricarea şi/sau inspecţia şi testarea finală a produselor sau a elementelor acestora sînt efectuate de o terţă parte, a metodelor de monitorizare a funcţionării eficace a sistemului calităţii şi, în special, tipul şi amploarea controalelor aplicate terţei părţi în cauză;
c) procedurilor de monitorizare şi verificare a proiectelor produselor şi, în special:
(i) a specificaţiilor de proiectare, inclusiv a standardelor care se aplică, şi o descriere a soluţiilor adoptate pentru a îndeplini cerinţele esenţiale aplicabile produselor, atunci cînd standardele menţionate la pct. 17 din prezentul Regulament nu sînt aplicate integral;
(ii) a tehnicilor de control şi verificare a proiectării, a proceselor şi acţiunilor sistematice ce urmează a fi utilizate în cadrul proiectării produselor;
(iii) a declaraţiei care indică dacă dispozitivul cuprinde sau nu, ca parte integrantă, o substanţă ori un derivat din sînge uman menţionată/menţionat la pct.11-15 din anexa nr. 1 la prezentul Regulament, precum şi datele referitoare la testele efectuate în această privinţă, necesare pentru a evalua securitatea, calitatea şi utilitatea acelei substanţe sau ale derivatului din sînge uman în cauză, conform scopului propus al dispozitivului;
(iv) a evaluării preclinice;
(v) a evaluării clinice menţionate în anexa nr. 7 la prezentul Regulament;
d) tehnicilor de control şi de asigurare a calităţii în faza de fabricaţie şi, în special:
(i) a proceselor şi procedurilor ce urmează a fi utilizate, în special cu privire la sterilizare, achiziţie de materiale şi la documentele relevante;
(ii) a procedurilor de identificare a produsului, întocmite şi actualizate din desene, specificaţii sau alte documente relevante, în fiecare fază a procesului de fabricaţie;
e) testelor şi verificărilor adecvate, care vor fi efectuate înainte, în timpul şi după procesul de fabricaţie, a frecvenţei acestora şi a echipamentului de testare folosit.
7. Fără a aduce atingere dispoziţiilor pct. 60, 61 şi 62 din prezentul Regulament, organismul recunoscut efectuează un audit al sistemului calităţii, pentru a stabili dacă acesta îndeplineşte cerinţele menţionate la pct. 5 şi 6 la prezenta anexă. Sistemele calităţii care folosesc standardele armonizate corespunzătoare sînt conforme cerinţelor sus-menţionate.
Echipa responsabilă de evaluare include cel puţin un membru care are experienţă în evaluarea tehnologiilor respective.
Procedura de evaluare include o inspecţie la sediul producătorului şi, în cazuri justificate în mod corespunzător, la sediile furnizorilor producătorului şi/sau ale subcontractanţilor, pentru a inspecta procesele de fabricaţie.
Decizia se comunică producătorului după inspecţia finală. Aceasta cuprinde concluziile activităţii de control şi o evaluare argumentată.
8. Producătorul informează organismul recunoscut, care a aprobat sistemul calităţii, cu privire la orice plan de modificare a sistemului calităţii. Organismul recunoscut evaluează modificările propuse şi verifică dacă sistemul calităţii astfel modificat mai respectă cerinţele menţionate la pct. 5 şi 6 din prezenta anexă; acesta face cunoscută decizia sa producătorului. Această decizie cuprinde concluziile activităţii de control şi o evaluare argumentată.
Secţiunea a II-a. Expertizarea proiectului produsului
9. Faţă de obligaţiile expuse în secţiunea I din prezenta anexă, producătorul înaintează organismului recunoscut cererea de examinare a dosarului de proiect referitor la produsul pe care intenţionează să îl fabrice şi care se înscrie în categoria menţionată la pct. 4 din prezenta anexă.10. Cererea descrie proiectarea, procesul de fabricaţie şi performanţele produsului respectiv şi include documentele necesare evaluării conformităţii produsului cu cerinţele prezentului Regulament, în special, cu cele prevăzute la pct. 5 şi pct.6 subpct.1) lit.c) şi d) din prezenta anexă.
Cererea include printre altele:
a) specificaţiile de proiectare, inclusiv standardele care au fost aplicate;
b) dovada din care să rezulte că standardele respective sînt corespunzătoare, mai ales în cazul în care standardele menţionate la pct. 17 din prezentul Regulament nu au fost aplicate integral. Această dovadă urmează să includă rezultatele încercărilor adecvate efectuate de către producător sau sub responsabilitatea acestuia;
c) declaraţia din care să rezulte dacă dispozitivul încorporează sau nu, ca parte integrantă, o substanţă de tipul celor menţionate la pct.11-15 din anexa nr.1 la prezentul Regulament, a cărei acţiune în asociere cu dispozitivul poate determina biodisponibilitatea sa, însoţită de date asupra investigaţiilor relevante realizate;
d) evaluarea clinică menţionată în anexa nr. 7 la prezentul Regulament;
e) proiectul broşurii cu instrucţiunile de utilizare.
11. Organismul recunoscut examinează solicitarea, iar dacă produsul este în conformitate cu prevederile relevante ale Regulamentului, eliberează solicitantului un certificat de examinare a proiectului. Organismul recunoscut poate cere ca solicitarea să fie completată cu teste sau cu probe suplimentare care să permită evaluarea conformităţii cu cerinţele prezentului Regulament. Certificatul cuprinde concluziile examinării, condiţiile de validitate ale acestuia, datele necesare identificării proiectului aprobat şi, după caz, descrierea scopului propus al produsului.
12. În cazul dispozitivelor menţionate la pct. 12 din anexa nr. 1 la prezentul Regulament, înainte de a lua o decizie, organismul recunoscut consultă, în ceea ce priveşte aspectele vizate la acel punct, Agenţia sau EMA. Avizul Agenţiei sau al EMA se emite în termen de 210 zile de la primirea unei documentaţii valide. Avizul ştiinţific al Agenţiei sau al EMA trebuie să fie inclus în documentaţia privind dispozitivul. La luarea deciziei, organismul recunoscut ia în considerare punctele de vedere exprimate cu ocazia acestei consultări. Acesta transmite decizia sa finală organismului competent implicat.
13. În cazul dispozitivelor menţionate în capitolul II şi pct.13 din anexa nr. 1 la prezentul Regulament, avizul ştiinţific al Agenţiei sau al EMA trebuie inclus în documentaţia privind dispozitivul. Avizul se emite în termen de 210 zile de la primirea unei documentaţii valide. La luarea deciziei, organismul recunoscut ia în considerare avizul Agenţiei sau al EMA. Organismul recunoscut poate să nu elibereze certificatul, în cazul în care avizul ştiinţific al Agenţiei sau al EMA este nefavorabil. Acesta transmite decizia sa finală către Agenţie sau EMA.
14. Solicitantul informează organismul recunoscut care a emis certificatul de examinare a proiectului despre orice modificare efectuată la proiectul aprobat. Pentru modificările efectuate la proiectul aprobat este necesară obţinerea unei aprobări suplimentare de la organismul recunoscut care a emis certificatul de examinare a proiectului, în cazul în care aceste modificări pot afecta conformitatea cu cerinţele esenţiale din prezentul Regulament sau condiţiile prescrise pentru utilizarea produsului. Această aprobare suplimentară se acordă sub forma unui addendum la certificatul de examinare a proiectului.
Secţiunea a III-a. Supravegherea
15. Scopul supravegherii este de a asigura că producătorul îndeplineşte obligaţiile impuse prin sistemul calităţii aprobat.16. Producătorul autorizează organismul recunoscut să efectueze toate inspecţiile necesare şi îi furnizează toate informaţiile, în special în legătură cu:
a) documentaţia privind sistemul calităţii;
b) datele prevăzute în acea parte a sistemului calităţii care se referă la proiect, cum ar fi rezultatele analizelor, calculelor, testelor, evaluarea preclinică, evaluarea clinică, planul de monitorizare clinică postvînzare şi rezultatele monitorizării clinice postvînzare, dacă este cazul;
c) datele prevăzute în partea sistemului calităţii care se referă la fabricaţie, cum ar fi: rapoartele referitoare la inspecţii, testele, standardizările/calibrările şi calificările personalului implicat.
17. Organismul recunoscut efectuează periodic inspecţiile şi evaluările necesare pentru a se asigura că producătorul aplică sistemul calităţii aprobat şi pune la dispoziţia producătorului un raport de evaluare.
18. În plus, organismul recunoscut poate face vizite neanunţate producătorului, punîndu-i acestuia la dispoziţie un raport de inspecţie.
Secţiunea a IV-a. Prevederile administrative
19. Timp de cel puţin 15 ani de la ultima dată de fabricaţie a produsului, producătorul sau reprezentantul său autorizat păstrează la dispoziţia Agenţiei:a) declaraţia de conformitate;
b) documentaţia prevăzută la pct. 4 lit. b), în special documentaţia, datele şi înregistrările menţionate în pct. 5 şi 6 din prezenta anexă;
c) amendamentele specificate la pct.8 din prezenta anexă;
d) documentaţia specificată la pct. 10 din prezenta anexă;
e) deciziile şi rapoartele organismului recunoscut, specificate la pct. 8, 11, 12, 13, 17 şi 18 din prezenta anexă.
20. La cerere, organismul recunoscut pune la dispoziţia celorlalte organisme recunoscute şi a Agenţiei toate datele relevante cu privire la aprobările sistemelor calităţii emise, respinse sau retrase.
21. După fabricarea fiecărui lot de dispozitive prevăzute la pct. 5 din prezentul Regulament, producătorul informează organismul recunoscut despre eliberarea lotului de dispozitive şi îi transmite certificatul oficial de eliberare a lotului de derivat din sînge uman utilizat în dispozitiv, certificat emis de un laborator de stat.
Anexa nr. 3
la Regulamentul privind condiţiile
de plasare pe piaţă a dispozitivelor
medicale implantabile active
la Regulamentul privind condiţiile
de plasare pe piaţă a dispozitivelor
medicale implantabile active
EXPERTIZAREA CE DE TIP
1. Examinarea CE de tip este procedura prin care un organism recunoscut constată şi certifică faptul că un exemplar reprezentativ din producţia avută în vedere îndeplineşte prevederile relevante din prezentul Regulament.2. Cererea pentru examinarea CE de tip se adresează de către producător sau de către reprezentantul său autorizat stabilit în Republica Moldova unui organism recunoscut.
Cererea cuprinde:
1) denumirea şi adresa producătorului şi a reprezentantului autorizat, dacă cererea este adresată de acesta din urmă;
2) declaraţia scrisă prin care se specifică faptul că o astfel de cerere nu a mai fost adresată nici unui alt organism recunoscut;
3) documentaţia descrisă la pct. 3 din prezenta anexă necesară pentru a permite evaluarea conformităţii unui eşantion reprezentativ din producţia respectivă (în continuare – tip), conform cerinţelor esenţiale din prezentul Regulament. Solicitantul asigură punerea unui tip la dispoziţia organismului recunoscut, iar acesta poate cere şi alte eşantioane, dacă este necesar.
3. Documentaţia urmează să asigure înţelegerea proiectării, fabricaţiei şi performanţelor dispozitivului şi cuprinde, în special, următoarele aspecte:
1) descrierea generală a tipului, inclusiv variantele avute în vedere, şi a utilizării (utilizărilor) prevăzute ale acestuia;
2) desenele de proiect, metodele de fabricaţie planificate, în special cu privire la sterilizare, precum şi diagramele de componente, subansambluri şi circuite;
3) descrierile şi explicaţiile necesare pentru înţelegerea desenelor şi diagramelor menţionate la subpct.2) din prezentul punct, precum şi descrierea funcţionării produsului;
4) lista standardelor menţionate la pct. 17 din prezentul Regulament, aplicate integral sau parţial, precum şi o descriere a soluţiilor adoptate pentru îndeplinirea cerinţelor esenţiale în cazul în care nu sînt aplicate standardele menţionate la pct. 17 din prezentul Regulament;
5) rezultatele calculelor de proiectare, analiza riscurilor, investigaţiile, testele tehnice efectuate;
6) declaraţia care indică dacă dispozitivul încorporează sau nu, ca parte integrantă, o substanţă ori un derivat din sînge uman, conform prevederilor de la pct.11-15 din anexa nr. 1 la prezentul Regulament, precum şi datele referitoare la testele efectuate în această privinţă, necesare pentru a evalua securitatea, calitatea şi utilitatea substanţei sau a derivatului din sînge uman, conform scopului propus al dispozitivului;
7) evaluarea preclinică;
8) evaluarea clinică menţionată în anexa nr. 7 la prezentul Regulament;
9) proiectul broşurii cu instrucţiunile de utilizare.
Secţiunea I. Organismul recunoscut
4. Organismul recunoscut:1) examinează şi evaluează documentaţia şi verifică dacă tipul este fabricat în conformitate cu această documentaţie; de asemenea, înregistrează elementele care au fost proiectate în conformitate cu prevederile aplicabile ale standardelor menţionate la pct. 17 din prezentul Regulament, precum şi elementele pentru care proiectarea nu se bazează pe prevederile acestor standarde;
2) efectuează sau solicită efectuarea inspecţiilor şi a încercărilor necesare pentru a verifica dacă soluţiile adoptate de producător îndeplinesc cerinţele esenţiale din prezentul Regulament, în cazul în care standardele menţionate la pct. 17 din prezentul Regulament nu au fost aplicate;
3) efectuează sau solicită efectuarea inspecţiilor şi a încercărilor necesare pentru a verifica dacă, în cazul în care producătorul a optat pentru aplicarea standardelor relevante, acestea au fost într-adevăr aplicate;
4) stabileşte de comun acord cu solicitantul locul unde se efectuează inspecţiile şi încercările necesare.
5. În cazul în care tipul este conform cu prevederile prezentului Regulament, organismul recunoscut eliberează solicitantului un certificat de examinare CE de tip. Certificatul cuprinde denumirea şi adresa producătorului, concluziile controlului, condiţiile de validitate şi datele necesare pentru identificarea tipului aprobat. Părţile relevante ale documentaţiei se anexează certificatului, iar o copie se păstrează de organismul recunoscut.
6. În cazul dispozitivelor menţionate la pct. 12 din anexa nr. 1 la prezentul Regulament, înainte de a lua o decizie, organismul recunoscut consultă Agenţia sau EMA în ceea ce priveşte aspectele vizate la acel punct.
Avizul Agenţiei sau al EMA se emite în termen de 210 zile de la primirea unei documentaţii valide. Avizul ştiinţific al Agenţiei sau al EMA trebuie inclus în documentaţia privind dispozitivul.
La luarea deciziei, organismul recunoscut ia în considerare punctele de vedere exprimate în aviz. Acesta transmite decizia sa finală organismului competent implicat.
7. În cazul dispozitivelor menţionate la pct.13 din anexa nr. 1 la Regulament, avizul ştiinţific al Agenţiei sau al EMA este inclus în documentaţia privind dispozitivul.
Avizul se emite în termen de 210 zile de la primirea unei documentaţii valide. La luarea deciziei, organismul recunoscut ia în considerare avizul Agenţiei sau al EMA.
Organismul recunoscut nu eliberează certificatul în cazul în care avizul ştiinţific al Agenţiei sau al EMA este nefavorabil. Acesta transmite decizia sa finală către Agenţie sau al EMA.
8. Solicitantul informează organismul recunoscut care a emis certificatul de examinare CE de tip cu privire la orice modificare efectuată asupra produsului aprobat.
9. Modificările asupra produsului aprobat trebuie să primească o altă aprobare din partea organismului recunoscut care a emis certificatul de examinare CE de tip, în cazul în care astfel de modificări pot afecta conformitatea cu cerinţele esenţiale sau condiţiile prevăzute pentru utilizarea produsului. Noua aprobare este eliberată, dacă este cazul, sub forma unui supliment la certificatul de examinare CE de tip iniţial.
Secţiunea a II-a. Prevederile administrative
10. Fiecare organism recunoscut urmează să comunice, la cerere, altor organisme recunoscute şi autorităţii competente toate informaţiile privind certificatele de examinare CE de tip şi suplimentele emise, respinse sau retrase.11. Alte organisme recunoscute pot obţine o copie de pe certificatele de examinare CE de tip şi/sau de pe suplimentele acestora. Anexele la certificate trebuie să fie accesibile pentru alte organisme recunoscute, la solicitarea justificată a acestora şi după informarea prealabilă a producătorului.
12. Producătorul sau reprezentantul său autorizat păstrează documentaţia tehnică şi copiile certificatelor de examinare CE de tip şi suplimentelor acestora pentru o perioadă de cel puţin 15 ani de la fabricarea ultimului produs.
Anexa nr. 4
la Regulamentul privind condiţiile
de plasare pe piaţă a dispozitivelor
medicale implantabile active
la Regulamentul privind condiţiile
de plasare pe piaţă a dispozitivelor
medicale implantabile active
VERIFICAREA UNITĂŢII DE PRODUS
1. Verificarea unităţii de produs este procedura prin care producătorul sau reprezentantul său autorizat cu sediul în Republica Moldova asigură şi declară că produsele care fac obiectul procedurii prevăzute la pct. 3 din prezenta anexă sînt conforme cu tipul descris în certificatul de examinare CE de tip şi îndeplinesc cerinţele aplicabile ale prezentului Regulament.2. Producătorul sau reprezentantul său autorizat cu sediul în Republica Moldova ia măsurile necesare pentru ca procesul de fabricaţie să asigure conformitatea produselor cu tipul descris în certificatul de examinare CE de tip şi cu cerinţele aplicabile din prezentul Regulament. Producătorul sau reprezentantul său autorizat cu sediul în Republica Moldova aplică marcajul de conformitate pe fiecare produs şi emite, în scris, o declaraţie de conformitate.
3. Înainte de începerea fabricaţiei, producătorul redactează documentele care definesc procesul de fabricaţie, în special privind sterilizarea, împreună cu toate prevederile de rutină prestabilite, pentru a asigura o producţie omogenă şi conformitatea produselor cu tipul descris în certificatul de examinare CE de tip şi cu cerinţele relevante din prezentul Regulament.
4. Producătorul se angajează să instituie şi să menţină la zi un sistem de supraveghere postvînzare, care să includă dispoziţiile menţionate în anexa nr. 7 la prezentul Regulament. Acest angajament include obligaţia producătorului de a comunica Agenţiei, imediat ce a luat cunoştinţă de acestea, următoarele evenimente:
1) orice deteriorare a caracteristicilor sau performanţelor dispozitivului, precum şi orice inexactitate din instrucţiunile de utilizare, care ar putea conduce sau au condus la decesul unui pacient ori la deteriorarea stării de sănătate al acestuia;
2) orice argument tehnic sau medical care a condus la retragerea de pe piaţă a unui dispozitiv de către producător.
5. Organismul recunoscut efectuează examinările şi încercările necesare pentru a verifica dacă produsul este conform cu cerinţele prezentului Regulament, prin examinarea şi testarea statistică a produselor, în conformitate cu prevederile pct. 6 din prezenta anexă. Producătorul autorizează organismul recunoscut să evalueze eficienţa măsurilor adoptate, potrivit prevederilor pct. 3 din prezenta anexă, prin audit, unde este cazul.
6. Verificarea statistică
1) Producătorii prezintă produsele fabricate sub formă de loturi omogene şi iau toate măsurile necesare astfel încît procesul de fabricaţie să asigure uniformitatea fiecărui lot de producţie realizat.
2) Se prelevă aleatoriu un eşantion din fiecare lot. Produsele din cadrul eşantionului se examinează individual şi se efectuează încercările corespunzătoare, definite în standardele menţionate la pct. 17 din prezentul Regulament, sau încercările echivalente pentru verificarea conformităţii produselor cu modelul descris în certificatul de examinare CE de tip, în scopul deciziei acceptării sau refuzării lotului de producţie.
3) Controlul statistic al produselor se bazează pe atribute şi/sau variabile, ceea ce implică un sistem de prelevare a eşantioanelor, cu caracteristici operaţionale, care să asigure un nivel ridicat de securitate şi performanţă în funcţie de nivelul tehnicii la momentul respectiv. Sistemele de prelevare a eşantioanelor se stabilesc în conformitate cu standardele armonizate menţionate la pct. 17 din prezentul Regulament, luînd în considerare specificul categoriilor de produse în discuţie.
4) În cazul în care loturile sînt acceptate, organismul recunoscut aplică sau impune aplicarea numărului său de identificare pe fiecare produs şi eliberează un certificat de conformitate privind încercările efectuate. Toate produsele din lot pot fi introduse pe piaţă, cu excepţia produselor neconforme din eşantionul examinat.
În cazul în care un lot este respins, organismul recunoscut ia măsurile necesare pentru a preveni introducerea pe piaţă a lotului respectiv. În eventualitatea respingerii frecvente a loturilor, organismul recunoscut suspendă verificarea statistică.
Producătorul poate, sub responsabilitatea organismului recunoscut, să aplice numărul de identificare al acestuia în timpul procesului de fabricaţie.
5) Producătorul sau reprezentantul său autorizat trebuie să asigure că este capabil de a furniza, la cerere, certificatele de conformitate emise de organismul recunoscut.
7. După fabricarea fiecărui lot de dispozitive prevăzute la pct. 5 din prezentul Regulament, producătorul informează organismul recunoscut despre eliberarea lotului de dispozitive şi îi transmite certificatul oficial de eliberare a lotului de derivat din sînge uman utilizat în dispozitiv, certificat emis de un laborator de stat.
Anexa nr. 5
la Regulamentul privind condiţiile
de plasare pe piaţă a dispozitivelor
medicale implantabile active
la Regulamentul privind condiţiile
de plasare pe piaţă a dispozitivelor
medicale implantabile active
DECLARAŢIE DE CONFORMITATE
privind asigurarea calităţii procesului de fabricaţie
1. Producătorul aplică sistemul calităţii aprobat pentru fabricaţie şi efectuează inspecţia finală a produselor respective conform celor specificate la pct. 3 din prezenta anexă, fiind subiectul supravegherii prevăzute la pct. 4 din prezenta anexă.privind asigurarea calităţii procesului de fabricaţie
2. Declaraţia de conformitate este o parte a procedurii prin care producătorul care îndeplineşte obligaţiile impuse la pct. 1 din prezenta anexă garantează şi declară că produsele respective sînt conforme cu tipul descris în certificatul de examinare CE de tip şi respectă prevederile prezentului Regulament, aplicabile acestora.
Producătorul sau reprezentantul său autorizat cu sediul în Republica Moldova aplică marcajul de conformitate potrivit prevederilor capitolului X din prezentul Regulament şi întocmeşte, în scris, o declaraţie de conformitate. Această declaraţie acoperă unul sau mai multe dispozitive identificate clar prin intermediul denumirii ori al codului produsului sau al altei referinţe lipsite de ambiguitate şi se păstrează de către producător. Marcajul de conformitate este însoţit de numărul de identificare al organismului recunoscut responsabil.
Secţiunea I. Sistemul calităţii
3. Producătorul înaintează o cerere de evaluare a sistemului calităţii la un organism recunoscut.Cererea cuprinde:
a) toate informaţiile adecvate referitoare la produsele pe care intenţionează să le fabrice;
b) documentaţia sistemului calităţii;
c) angajamentul producătorului de a îndeplini obligaţiile ce decurg din sistemul calităţii aprobat;
d) angajamentul producătorului de a menţine sistemul calităţii aprobat în aşa fel încît acesta să rămînă adecvat şi eficace;
e) documentaţia tehnică privind tipul aprobat şi o copie a certificatului de examinare CE de tip, după caz;
f) angajamentul producătorului de a institui şi de a menţine actualizat un sistem de supraveghere postvînzare, care să includă dispoziţiile prevăzute în anexa nr. 7 la prezentul Regulament. Acest angajament include obligaţia asumată de către producător de a comunica Agenţiei următoarele incidente imediat ce a luat cunoştinţă despre acestea:
(i) orice deteriorare a caracteristicilor sau performanţelor dispozitivului, precum şi orice inexactitate din instrucţiunile de utilizare, care ar putea duce sau au dus la decesul unui pacient ori la deteriorarea stării sale de sănătate;
(ii) orice argument tehnic sau medical care a condus la retragerea unui dispozitiv de pe piaţă de către producător.
4. Aplicarea sistemului calităţii asigură că produsele sînt conforme cu tipul descris în certificatul de examinare CE de tip.
5. Toate elementele, cerinţele şi dispoziţiile adoptate de producător pentru sistemul calităţii sînt documentate în mod sistematic şi ordonat sub formă de proceduri şi politici scrise. Această documentaţie a sistemului calităţii trebuie să permită o interpretare uniformă a politicilor calităţii şi a procedurilor, cum ar fi: programele calităţii, planurile calităţii, manualele calităţii şi înregistrările calităţii.
Documentaţia include, în special, o descriere adecvată a:
a) obiectivelor producătorului privind calitatea;
b) organizării afacerilor, în special:
(i) a structurilor organizatorice, responsabilităţilor personalului de conducere şi a autorităţii acestuia, în cazul în care este vizată fabricaţia produselor;
(ii) a metodelor de monitorizare a funcţionării eficiente a sistemului calităţii, în special capacitatea acestuia de a determina calitatea dorită a produselor, inclusiv controlul produselor care nu sînt conforme;
(iii) în cazul în care fabricarea şi/sau inspecţia şi testarea finală a produselor sau a elementor acestora sînt efectuate de o terţă parte, a metodelor de monitorizare a funcţionării eficace a sistemului calităţii, în special tipul şi amploarea controalelor aplicate terţei părţi în cauză;
c) tehnicilor de control şi de asigurare a calităţii în faza de fabricaţie, în special:
(i) a proceselor şi procedurilor ce urmează a fi utilizate, în special, cu privire la sterilizare, achiziţie de materiale şi documentele relevante;
(ii) a procedurilor de identificare a produsului întocmite şi actualizate din desene, specificaţii sau alte documente relevante, în fiecare fază a procesului de fabricaţie;
d) testelor şi verificărilor adecvate, efectuate înainte, în timpul şi după procesul de fabricaţie, a frecvenţei acestora şi a echipamentului de testare folosit.
6. Fără a aduce atingere dispoziţiilor pct. 58 şi 59 din prezentul Regulament, organismul recunoscut efectuează un audit al sistemului calităţii pentru a stabili dacă acesta îndeplineşte cerinţele menţionate la pct. 4 şi 5 din prezenta anexă. Se consideră că sistemele calităţii care folosesc standardele armonizate corespunzătoare sînt conforme acestor cerinţe.
Echipa responsabilă de evaluare include cel puţin un membru care are experienţă în evaluarea tehnologiilor respective. Procedura de evaluare include o inspecţie la sediul producătorului.
Decizia se comunică producătorului după inspecţia finală. Aceasta cuprinde concluziile activităţii de control şi o evaluare motivată.
7. Producătorul informează organismul recunoscut care a aprobat sistemul calităţii cu privire la orice plan de modificare a sistemului calităţii. Organismul recunoscut evaluează modificările propuse şi verifică dacă sistemul calităţii astfel modificat mai respectă cerinţele menţionate la pct. 4 şi 5 din prezenta anexă; acesta face cunoscută decizia sa producătorului. Această decizie cuprinde concluziile activităţii de control şi o evaluare argumentată.
Secţiunea a II-a. Supravegherea
8. Scopul supravegherii este să asigure că producătorul îndeplineşte obligaţiile impuse prin sistemul calităţii aprobat.9. Producătorul autorizează organismul recunoscut să efectueze toate inspecţiile necesare şi îi furnizează toate informaţiile necesare, în special în legătură cu:
a) documentaţia privind sistemul calităţii;
b) documentaţia tehnică;
c) datele prevăzute în sistemul calităţii referitoare la fabricaţie, cum ar fi: rapoartele de inspecţie, testele, standardizarea/calibrarea şi calificările personalului
implicat etc.
10. Organismul recunoscut efectuează periodic inspecţiile şi evaluările necesare pentru a se asigura că producătorul aplică sistemul calităţii aprobat şi pune la dispoziţia producătorului un raport de evaluare.
11. În plus, organismul recunoscut, dacă este necesar, face vizite neanunţate producătorului, punîndu-i acestuia la dispoziţie un raport de inspecţie.
12. Organismul recunoscut comunică celorlalte organisme recunoscute toate datele relevante referitoare la aprobările privind sistemele de calitate emise, respinse sau retrase.
13. După fabricarea fiecărui lot de dispozitive prevăzute la pct. 5 din prezentul Regulament, producătorul informează organismul recunoscut despre eliberarea lotului de dispozitive şi îi transmite certificatul oficial de eliberare a lotului de derivat din sînge uman utilizat în dispozitiv, certificat emis de un laborator de stat sau de un laborator desemnat de stat.
Anexa nr. 6
la Regulamentul privind condiţiile
de plasare pe piaţă a dispozitivelor
medicale implantabile active
la Regulamentul privind condiţiile
de plasare pe piaţă a dispozitivelor
medicale implantabile active
DECLARAŢIA
privind dispozitivele destinate unor scopuri speciale
1. Producătorul sau reprezentantul său autorizat cu sediul în Republica Moldova întocmeşte declaraţia pentru dispozitivele fabricate la comandă sau pentru dispozitivele destinate investigaţiilor clinice, care cuprinde elementele prevăzute la pct. 2 din prezenta anexă.privind dispozitivele destinate unor scopuri speciale
2. Declaraţia cuprinde următoarele informaţii:
1) pentru dispozitivele fabricate la comandă:
a) denumirea şi adresa producătorului;
b) informaţiile necesare pentru identificarea produsului în cauză;
c) o declaraţie din care să rezulte că dispozitivul este destinat pentru a fi utilizat în exclusivitate de către un anumit pacient, menţionîndu-se numele pacientului;
d) numele practicianului medical calificat în mod corespunzător care a făcut prescripţia şi, după caz, numele clinicii implicate;
e) caracteristicile specifice ale produsului prezentate de prescripţia medicală;
f) o declaraţie din care să rezulte că dispozitivul respectiv satisface cerinţele esenţiale prevăzute în anexa nr. 1 la prezentul Regulament şi care, după caz, să indice care dintre cerinţele esenţiale nu au fost îndeplinite integral, împreună cu motivaţia;
2) pentru dispozitivele destinate investigaţiilor clinice prevăzute în anexa nr. 7 la prezentul Regulament:
a) datele care să permită identificarea dispozitivelor respective;
b) planul investigaţiei clinice;
c) broşura investigatorului;
d) confirmarea asigurării subiecţilor;
e) documentele utilizate pentru obţinerea consimţămîntului în cunoştinţă de cauză;
f) o declaraţie care indică dacă dispozitivul încorporează sau nu, ca parte integrantă, o substanţă ori un derivat din sînge uman prevăzută/prevăzut la pct. 11-15 din anexa nr. 1 la prezentul Regulament;
g) avizul comisiei de etică implicate şi detaliile privind aspectele abordate în avizul său;
h) numele practicianului medical calificat în mod corespunzător sau al altei persoane autorizate şi al instituţiei responsabile pentru investigaţii;
i) locul şi data începerii investigaţiilor şi durata programată pentru acestea;
j) declaraţia din care să rezulte că dispozitivul respectiv este conform cu cerinţele esenţiale, prevăzute în anexa nr. 1 la prezentul Regulament, excluzînd aspectele ce constituie obiectul investigaţiilor, şi că, în ceea ce priveşte aceste aspecte, s-au luat toate măsurile de precauţie pentru a proteja sănătatea şi securitatea pacientului.
3. Producătorul se angajează să pună la dispoziţia Agenţiei următoarele:
1) pentru dispozitivele fabricate la comandă, documentaţia care indică amplasamentul (amplasamentele) de producţie şi care permite înţelegerea proiectului, a fabricaţiei şi a performanţelor produsului, inclusiv performanţele urmărite, pentru a permite evaluarea conformităţii cu cerinţele prezentului Regulament.
Producătorul ia toate măsurile necesare pentru ca procesul de fabricaţie să asigure că produsele fabricate sînt conforme cu documentaţia menţionată mai sus;
2) pentru dispozitivele destinate investigaţiilor clinice, documentaţia cuprinde, de asemenea:
a) descrierea generală a produsului şi a utilizărilor prevăzute;
b) desenele de proiect, metodele de fabricaţie, în special în ceea ce priveşte sterilizarea, şi diagramele de componente, subansamblurile, circuitele;
c) descrierile şi explicaţiile necesare pentru înţelegerea respectivelor desene şi diagrame şi a funcţionării produsului;
d) rezultatele analizei riscurilor şi o listă cuprinzînd standardele prevăzute la pct. 17 din prezentul Regulament, aplicate integral sau în parte, precum şi o descriere a soluţiilor adoptate pentru îndeplinirea cerinţelor esenţiale din prezentul Regulament, în cazul în care standardele menţionate la pct. 17 din prezentul Regulament nu au fost aplicate;
e) în cazul în care dispozitivul încorporează, ca parte integrantă, o substanţă sau un derivat din sînge uman menţionată/menţionat la pct. 11-15 din anexa nr. 1 la prezentul Regulament, datele referitoare la testele efectuate în această privinţă, necesare pentru a evalua securitatea, calitatea şi utilitatea substanţei sau a derivatului de sînge uman, conform scopului propus al dispozitivului;
f) rezultatele calculelor proiectului, verificărilor şi testelor tehnice executate.
Producătorul ia toate măsurile necesare pentru ca procesul de fabricaţie să asigure că produsele fabricate sînt în conformitate cu documentaţia menţionată la pct. 3 subpct. 1) şi subpct. 2) lit. a)-f) din prezenta anexă.
Producătorul, dacă este necesar, poate autoriza evaluarea eficienţei acestor măsuri prin audit.
4. Informaţiile incluse în declaraţiile care intră sub incidenţa prezentei anexe se păstrează pentru o perioadă de cel puţin 15 ani de la data fabricaţiei ultimului produs.
5. Pentru dispozitivele fabricate la comandă, producătorul se angajează să revizuiască şi să documenteze experienţa acumulată după încheierea fazei de producţie, inclusiv dispoziţiile menţionate în anexa nr. 7 la prezentul Regulament, şi să implementeze mijloacele adecvate pentru aplicarea oricăror măsuri corective necesare. Acest angajament include obligaţia producătorului de a informa autorităţile competente cu privire la următoarele incidente imediat ce a aflat de existenţa lor, precum şi la măsurile corective relevante:
1) orice funcţionare defectuoasă sau deteriorare a caracteristicilor şi/sau a performanţelor unui dispozitiv, precum şi orice caz de inadecvare a etichetării sau a instrucţiunilor de utilizare, care pot să conducă sau au condus la decesul ori la deteriorarea severă a stării de sănătate a unui pacient sau utilizator;
2) orice cauză de ordin tehnic sau medical legată de caracteristicile sau performanţele unui dispozitiv, care, din motivele menţionate la subp.1) din prezentul punct, conduce la retragerea sistematică de pe piaţă de către producător a dispozitivelor de acelaşi tip.
Anexa nr. 7
la Regulamentul privind condiţiile
de plasare pe piaţă a dispozitivelor
medicale implantabile active
la Regulamentul privind condiţiile
de plasare pe piaţă a dispozitivelor
medicale implantabile active
INVESTIGAŢIA CLINICĂ
Secţiunea I. Prevederi generale
1. Ca regulă generală, confirmarea conformităţii cu cerinţele privind caracteristicile şi performanţele prevăzute la pct. 1 şi 2 din anexa nr. 1 la prezentul Regulament, în condiţii normale de utilizare a dispozitivului, şi evaluarea efectelor secundare şi a acceptabilităţii raportului beneficii/riscuri menţionat la pct. 5 din anexa nr. 1 la prezentul Regulament trebuie să se bazeze pe date clinice. Evaluarea acestor date (în continuare – evaluare clinică), respectînd, dacă este cazul, eventualele standarde armonizate relevante, urmează o procedură definită şi sigură din punct de vedere metodologic, bazată pe:Secţiunea I. Prevederi generale
a) fie o evaluare critică a literaturii ştiinţifice, curent disponibile, cu privire la securitatea, performanţele, caracteristicile proiectului şi scopul propus al dispozitivului, în care:
(i) se demonstrează echivalenţa dispozitivului cu dispozitivul la care fac referire datele; şi
(ii) datele demonstrează în mod adecvat conformitatea cu cerinţele esenţiale relevante;
b) fie o evaluare critică a rezultatelor tuturor investigaţiilor clinice efectuate;
c) fie o evaluare critică a datelor clinice combinate prevăzute la lit. a şi b din prezentul subpunct.
2. Se efectuează investigaţii clinice, cu excepţia cazurilor în care se justifică utilizarea datelor clinice existente.
3. Pentru evaluarea clinică şi rezultatul său se prezintă documente justificative. Documentaţia tehnică a dispozitivului include şi/sau face trimitere la documentaţia clinică în cauză.
4. Evaluarea clinică şi documentaţia aferentă se actualizează activ cu datele obţinute în cursul supravegherii după introducerea pe piaţă. În cazul în care se constată că supravegherea clinică după introducerea pe piaţă, ca parte integrantă a planului de supraveghere a dispozitivului după introducerea pe piaţă, nu este necesară, acest lucru se justifică şi se documentează în mod adecvat.
5. În cazul în care se constată că demonstrarea conformităţii cu cerinţele esenţiale pe baza datelor clinice nu este adecvată, se furnizează o justificare corespunzătoare a acestei excluderi, pe baza rezultatelor gestionării riscurilor şi luînd în considerare caracteristicile specifice ale interacţiunii dintre dispozitiv şi organismul uman, performanţele clinice prevăzute şi cererile producătorului. În cazul în care demonstrarea conformităţii cu cerinţele esenţiale se bazează exclusiv pe evaluarea performanţelor, teste pe banc şi evaluare preclinică, este necesar să se demonstreze în mod corespunzător că această metodă este adecvată.
6. Toate datele rămîn confidenţiale, în conformitate cu prevederile pct. 63 şi 64 din prezentul Regulament.
Secţiunea a II-a. Investigaţia clinică
1. Obiectivele investigaţiei clinice sînt:1) să verifice dacă, în condiţii normale de utilizare, performanţele dispozitivului sînt în conformitate cu cele prevăzute în pct. 2 din anexa nr. 1 la prezentul Regulament;
2) să determine orice efecte secundare nedorite, în condiţii normale de utilizare, şi să evalueze dacă acestea constituie riscuri acceptabile în raport cu performanţele preconizate ale dispozitivului.
2. Consideraţii etice
Investigaţiile clinice se efectuează în conformitate cu Declaraţia de la Helsinki, aprobată la cea de-a 18-a Adunare Medicală Mondială de la Helsinki, Finlanda, în 1964, modificată la cea de-a 29-a Adunare Medicală Mondială de la Tokio, Japonia, în 1975, şi la cea de-a 35-a Adunare Medicală Mondială de la Veneţia, Italia, în 1983. Este obligatoriu ca toate măsurile cu privire la protecţia subiecţilor umani să fie realizate în spiritul Declaraţiei de la Helsinki. Aceasta include toate etapele investigaţiei clinice, de la prima considerare cu privire la necesitatea şi justificarea studiului pînă la publicarea rezultatelor.
3. Metode:
1) Investigaţiile clinice se efectuează în conformitate cu un plan de investigaţie adecvat, conform nivelului ştiinţific al momentului, astfel definit încît să confirme sau să respingă cele invocate de producător cu privire la dispozitiv; investigaţiile urmează să includă un număr corespunzător de observaţii pentru a garanta validitatea ştiinţifică a concluziilor.
2) Procedurile folosite pentru efectuarea investigaţiilor urmează să fie adecvate dispozitivului destinat examinaării
3) Investigaţiile clinice se efectuează în circumstanţe echivalente condiţiilor normale de utilizare a dispozitivului.
4) Se examinează toate caracteristicile corespunzătoare, inclusiv cele privind siguranţa şi performanţele dispozitivului, precum şi efectele sale asupra pacienţilor.
5) Toate incidentele adverse grave urmează să fie înregistrate complet şi notificate de îndată tuturor autorităţilor competente ale statelor în care are loc investigaţia clinică.
6) Investigaţiile se efectuează sub responsabilitatea unui practician medical calificat în mod corespunzător sau a unei persoane autorizate, într-un mediu adecvat.
Specialistul medical beneficiază de acces la datele tehnice referitoare la dispozitiv.
7) Raportul scris, semnat de către medicul specialist responsabil, cuprinde o evaluare critică a tuturor datelor obţinute în timpul investigaţiei clinice.
Anexa nr.8
la Regulamentul privind condiţiile
de plasare pe piaţă a dispozitivelor
medicale implantabile active
la Regulamentul privind condiţiile
de plasare pe piaţă a dispozitivelor
medicale implantabile active
CRITERIILE MINIME
OBLIGATORII DE RECUNOAŞTERE A ORGANISMELOR
DE EVALUARE A CONFORMITĂŢII
1. Organismul recunoscut, conducătorul acestuia şi personalul responsabil pentru operaţiunile de evaluare şi verificare nu poate să fie proiectantul, producătorul, furnizorul sau instalatorul dispozitivelor pe care le controlează şi nici reprezentantul autorizat al vreuneia dintre aceste părţi. Aceştia nu pot fi direct implicaţi în proiectarea, fabricarea, comercializarea sau întreţinerea dispozitivelor şi nici nu pot reprezenta părţile angajate în aceste activităţi. Prin aceasta nu se exclude posibilitatea unui schimb de informaţii tehnice între producător şi organism.OBLIGATORII DE RECUNOAŞTERE A ORGANISMELOR
DE EVALUARE A CONFORMITĂŢII
2. Organismul recunoscut şi personalul său trebuie să efectueze operaţiunile de evaluare şi verificare la cel mai înalt standard de integritate profesională şi competenţă tehnică şi trebuie să fie în afara oricăror presiuni sau influenţe, în special de natură financiară, care ar putea influenţa decizia acestora sau rezultatele inspecţiei, în special din partea persoanelor ori a grupurilor de persoane interesate de rezultatul verificărilor.
3. Organismul recunoscut urmează să realizeze toate sarcinile prevăzute în anexele nr. 2–5 la prezentul Regulament, pentru care a fost recunoscut, indiferent dacă aceste sarcini sînt executate de organismul respectiv sau sub responsabilitatea sa. În special, trebuie să aibă la dispoziţie personalul necesar şi să posede facilităţile necesare pentru îndeplinirea corespunzătoare a sarcinilor tehnice şi administrative impuse de evaluare şi verificare.
Organismul recunoscut trebuie, de asemenea, să aibă acces la echipamentele necesare pentru verificările cerute.
4. Personalul responsabil de operaţiunile de control deţin:
1) pregătire profesională temeinică, care să acopere toate operaţiile de evaluare şi verificare pentru care a fost recunoscut organismul;
2) cunoştinţe corespunzătoare privind cerinţele operaţiunilor de control pe care le efectuează şi experienţă corespunzătoare în acest domeniu;
3) capacitatea necesară pentru întocmirea certificatelor, înregistrărilor şi rapoartelor, pentru a demonstra că s-au efectuat respectivele operaţiuni de control.
5. Imparţialitatea personalului care realizează inspecţia trebuie să fie garantată. Remuneraţia acestuia nu influenţează şi nu depinde de numărul inspecţiilor efectuate şi nici de rezultatul acestor inspecţii.
6. Organismul recunoscut trebuie să încheie o asigurare de răspundere civilă, cu excepţia cazului în care această răspundere este asigurată de către stat prin lege.
7. Personalul organismului recunoscut este obligat să păstreze secretul profesional cu privire la toate informaţiile obţinute pe durata sarcinilor ce-i revin prin prezentul Regulament sau prin orice prevederi ale legislaţiei în vigoare. Personalul organismului nu are obligaţia păstrării secretului profesional faţă de Agenţie.
Anexa nr. 9
la Regulamentul privind condiţiile
de plasare pe piaţă a dispozitivelor
medicale implantabile active
la Regulamentul privind condiţiile
de plasare pe piaţă a dispozitivelor
medicale implantabile active
MARCAJUL DE CONFORMITATE
1. Marca naţională de conformitate SM se aplică produselor industriale (în continuare – produse) din domeniul reglementat şi denotă faptul că producătorul sau reprezentantul său cu sediul în Republica Moldova (în continuare – producătorul sau reprezentantul său) care a aplicat sau care răspunde de aplicarea mărcii respective a verificat conformitatea produsului cu toate cerinţele esenţiale prevăzute în reglementările tehnice aplicabile acestuia şi că produsul a fost supus procedurilor de evaluare a conformităţii, prevăzute de reglementarea tehnică respectivă.2. Pentru produsele care fac obiectul mai multor reglementări tehnice ce prevăd aplicarea mărcii naţionale de conformitate SM, marca respectivă semnifică faptul că produsele în cauză sînt conforme cu prevederile tuturor reglementărilor tehnice aplicabile.
3. Marca naţională de conformitate SM este formată din literele S şi M, care simbolizează „securitate conform cerinţelor esenţiale” şi, respectiv, „Moldova”. Simbolul grafic al mărcii naţionale de conformitate SM este prezentat în figura 1.
4. Dimensiunile mărcii naţionale de conformitate SM trebuie să corespundă întocmai celor specificate în figura 2.
5. În cazul în care marca naţională de conformitate SM urmează a fi mărită sau micşorată, se vor respecta dimensiunile specificate în figura 2.
6. Marca naţională de conformitate SM se execută în alb-negru sau într-o singură culoare, în contrast cu fondul.
7. Pe orice produs din domeniul reglementat va fi aplicată marca naţională de conformitate SM.
8. Marca naţională de conformitate SM se execută prin orice procedeu tehnologic care asigură obţinerea unei imagini clare şi durabile a ei pe toată perioada de utilizare a produselor respective marcate.
9. Marca naţională de conformitate SM este însoţită de numărul de identificare al organismului de evaluare a conformităţii recunoscut, care a fost antrenat în faza de evaluare respectivă, conform prevederilor reglementărilor tehnice aplicabile. Numărul de identificare al organismului de evaluare a conformităţii desemnat se scrie la distanţa de 5% din înălţimea mărcii, sub desenul grafic al acesteia, simetric faţă de axa verticală, cu înălţimea literelor (cifrelor) pînă la 15% din înălţimea mărcii.
10. Marca naţională de conformitate SM şi numărul de identificare al organismului respectiv de evaluare a conformităţii desemnat se aplică de către producător sau de către reprezentantul său.
11. Marca naţională de conformitate SM este utilizată de către producător sau de către reprezentantul său în mod gratuit.
12. Pe un produs, concomitent cu marca naţională de conformitate SM, pot fi aplicate mărci diferite (de exemplu, mărci ce indică conformitatea cu standardele naţionale sau europene sau cu alte reglementări), cu condiţia ca aceste mărci să nu poată fi confundate cu marca naţională de conformitate SM. Aceste mărci pot fi aplicate cu condiţia ca lizibilitatea şi vizibilitatea mărcii naţionale de conformitate SM să nu fie afectate.
13. Dacă organul de supraveghere şi control stabileşte că marca naţională de conformitate SM a fost aplicată neadecvat, producătorul, reprezentantul său sau (în mod excepţional dacă reglementarea tehnică aplicabilă prevede astfel) persoana responsabilă de plasarea produsului respectiv pe piaţă are obligaţia să înlăture neconformităţile respective.
14. În cazul în care neconformitatea nu este înlăturată, organul de supraveghere şi control restricţionează sau interzice distribuirea sub orice formă a produsului respectiv ori asigură retragerea de pe piaţă a acestuia, în conformitate cu prevederile legislaţiei în vigoare.