Внутренний номер: 331278
Varianta în limba de stat

Республика Молдова
от 06.04.2009
и интерпретации результатов в области ветеринарной санитарии
ПП51 от 16.01.13, МО15-17/22.01.13 ст.89
В соответствии с положениями Закона № 221-XVI от 19 октября 2007 года о ветеринарно-санитарной деятельности (Официальный монитор Республики Молдова, 2008 г., № 51-54, ст. 153), Закона № 1513-XII от 16 июня 1993 года о санитарно-эпидемиологическом обеспечении населения (повторное опубликование: Официальный монитор Республики Молдова, 2003 г., № 60-61, ст. 259), Закона № 78-XV от 18 марта 2004 года о пищевых продуктах (Официальный монитор Республики Молдова, 2004 г., № 83-87, ст. 431), в целях исключения для здоровья общества рисков, исходящих от некоторых веществ, медикаментов ветеринарного назначения и их остатков в живых животных и продуктах животного происхождения, а также обеспечения потребителя безопасными и безвредными пищевыми продуктами путем применения передовых методов обнаружения вредных веществ и правильной интерпретации полученных результатов Правительство ПОСТАНОВЛЯЕТ:
1. Утвердить Регламент применения методов испытаний и интерпретации результатов в области ветеринарной санитарии (прилагается).
2. Контроль за исполнением настоящего Постановления возложить на Министерство сельского хозяйства и пищевой промышленности.
Премьер-министр Зинаида ГРЕЧАНЫЙ
Контрассигнует:
министр сельского хозяйства и
пищевой промышленности Анатолие Городенко
Постановлением Правительства
№ 265 от 6 апреля 2009 г.
применения методов испытаний и интерпретации
результатов в области ветеринарной санитарии
2. Настоящий Регламент не применяется к веществам, для которых установлены специальные нормы, предусмотренные национальным законодательством.
3. Положения настоящего Регламента перенимают положения Решения Комиссии Европейских Сообществ № 2002/657 от 14 августа 2002 г., устанавливающего нормы по применению Директивы № 96/23/CE Совета по функционированию методов исследования и интерпретации результатов (Официальный журнал Европейского Союза, L 221, 17.08.2002, стр. 8).
4. В настоящем Регламенте используются определения, касающиеся мер по надзору и контролю некоторых веществ, их остатков и остатков препаратов ветеринарного назначения у живых животных и в продуктах животного происхождения, а также перечисленных в приложении к настоящему Регламенту.
5. Национальное агентство по безопасности пищевых продуктов (в дальнейшем – Агентство) должно принимать все меры, чтобы официальные пробы, отобранные официальными и/или уполномоченными ветеринарными врачами, исследовались методами, которые:
[Пкт.5 абз. изменен ПП51 от 16.01.13, МО15-17/22.01.13 ст.89]
a) зарегистрированы в испытательных инструкциях в соответствии со стандартом ISO 5725 - 4;
b) соответствуют разделу 2 приложения к настоящему Регламенту;
c) признаны (валидированы) в соответствии с процедурами, предусмотренными в разделе 3 приложения к настоящему Регламенту.
6. Аналитические методы должны отвечать требуемым минимальным пределам функционирования (MRPL).
Для веществ, для которых не установлены разрешенные пределы, национальный компетентный орган предлагает, на основе научно-технического прогресса и установления некоторых норм на международном уровне, дополнения в целях прогрессивного установления требуемых минимальных пределов функционирования (MRPL), которые будут применяться к используемым методам испытаний.
7. Национальный компетентный орган должен обеспечить качество результатов испытаний проб, в частности, путем контрольных исследований и/или эталонных результатов в соответствии с SM SR EN ISO/CEI 17025:2006.
8. Результат испытания считается несоответствующим в случае, если превышается предел чувствительности метода подтверждения для исследуемого аналита.
9. При установлении разрешенного предела для определенного вещества пределом чувствительности считается концентрация, от которой возможно решить со статистической достоверностью в 1 – α, что действительно превышен разрешенный предел.
10. Если не установлен разрешенный предел для определенного вещества, пределом чувствительности считается минимальный уровень концентрации, при котором метод может доказывать со статистической достоверностью в 1 – α присутствие исследуемого аналита.
11. Для неразрешенных веществ и веществ с анаболическим эффектом, стильбенов, производных стильбенов, их солей из эфиров, антитироидов, стероидов, лактонов резорциновой кислоты, включая зеранол, бета-аганисты, активные фармакологические вещества, для которых невозможно предусмотреть максимальные пределы для продуктов животного происхождения (в дальнейшем – вещества группы А), ошибка α должна быть равна или меньше 1%.
Для остальных веществ ошибка α равна или меньше 5%.
12. Республиканскому ветеринарно-диагностическому центру, уполномоченному проводить эти испытания, предоставляется срок, не превышающий 3-х лет, для приведения в соответствие с положениями настоящего Регламента.
13. В случае, когда внедрение некоторых методов испытаний в соответствии с положениями настоящего Регламента являются нерациональным или невозможным, лаборатории, обязанные проводить эти испытания, должны заключать через посредство национального компетентного органа договоры на проведение испытаний для подтверждения результатов с одной из референтных лабораторий из других государств.
к Регламенту применения методов
испытаний и интерпретации
результатов в области ветеринарной
санитарии
применяемые к методам испытаний
точность измерения – степень соответствия между результатом одного измерения и значением условно правильным значением измеренной величины;
ошибка альфа (α) – вероятность, что исследуемая проба соответствует, даже если полученное измерение не соответствует («решение принятое на ошибочно соответствующем результате»);
аналит – вещество, которое должно быть обнаружено, идентифицировано и/или количественно определено, и производные, появляющиеся во время анализа;
ошибка бета (β) – вероятность, что исследуемая проба достоверно не соответствует, даже если полученное измерение соответствует («решение, основанное на ложно соответствующем результате»);
систематическая погрешность – разница между средней арифметической результатов бесконечного числа измерений того же образца, выполненных в условиях повторяемости и реального значения образца;
эталон – средство измерения, предназначенное для определения, выполнения, сохранения или воспроизводства единицы или одного или более значений того же измерения, которое будет служить в качестве референтного;
материал референтный сертифицированный (МРС) – референтный материал, сопровождаемый сертификатом, значение (одно или несколько) свойства (свойств) которого сертифицировано процедурой, обеспечивающей прослеживаемость правильного выполнения единицы (единиц), у которой выражены значения свойства (свойств) и для которого каждое сертифицированное значение сопровождается неуверенностью с указанным уровнем достоверности;
материал референтный – средство измерения, материал или вещество, значение (одно или несколько) свойства (свойств) которого достаточно однородное и хорошо установлено для того, чтобы быть использованным для проверки и/или эталонирования средства измерения, для проверки процедуры законного измерения или присвоения значения материалам или веществам;
хроматография комбинированная – метод, при котором экстракт делится на две части. Одна часть подлежит непосредственно хроматографическому исследованию. Вторая часть смешивается с эталоном аналита, затем подлежит хроматографическому исследованию. Количество добавленного эталонного аналита должно быть приблизительно равно количеству определенного аналита в экстракте. Этот метод позволяет улучшить идентификацию аналита путем хроматографического исследования, в частности, если при этом невозможно использовать соответствующий внутренний стандарт;
совместное исследование – анализ той же пробы, тем же методом в целях определения характеристик применения метода. Исследование покрывает случайную ошибку измерения и систематическую ошибку лаборатории;
подтверждающий метод – метод, который обеспечивает полную или дополнительную информацию, позволяющую недвусмысленно идентифицировать вещество и, при необходимости, определить его количество на уровне, представляющем интерес;
предел определения (CCα) – предел, который и выше которого позволяется заключить с вероятностью ошибки α, что проба не соответствует;
способность обнаружения (CCβ) – наименьшее содержание вещества, которое может быть обнаружено, идентифицировано и/или количественно определено в пробе с вероятностью ошибки β. Для веществ, для которых не установлен допустимый уровень, способностью обнаружения является самая низкая концентрация, при которой метод способен обнаружить действительно загрязненные пробы со статистической уверенностью β. Для веществ, для которых установлен допустимый уровень, способностью обнаружения является концентрация, при которой метод может обнаружить допустимые уровни со статистической достоверностью 1 – β;
обогащенный материал пробы – проба, к которой добавляется известное количество аналита, который следует обнаружить;
межлабораторное изучение (сравнительное) – организация, выполнение и оценка методов испытаний на одной и той же пробе в двух или более лабораториях в соответствии с заранее установленными условиями в целях определения порядка применения метода. В зависимости от цели, изучение может быть классифицировано как совместное изучение или тест опытности;
внутренний эталон – вещество, которое не содержится в пробе, но имеет физико-химические свойства, схожие с таковыми аналита, который должен быть идентифицирован, которое добавляется к каждой пробе, а также к каждому эталону;
официальная проба – количество биологического материала или продукта животного происхождения, отобранного из партии официальным или уполномоченным ветеринарным врачом в рамках программы по ветеринарно-санитарному надзору и контролю, результаты исследования которой служат основой для принятия решения лицом, отбиравшим данную пробу;
лабораторная проба – проба, подготовленная к отправке в лабораторию и предназначенная для исследования или испытания;
учтенный уровень – концентрация вещества или аналита в пробе, которая является достоверной для определения его соответствия законодательству;
требуемый минимальный предел функционирования (MRPL) – минимальное количество аналита в пробе, которое необходимо, по меньшей мере, выявить и подтвердить. Предназначены для гармонизации аналитических способностей методов, которые применяются к веществам, для которых не установлены допустимые уровни;
параметры работы – функциональное качество, которое может быть присвоено аналитическому методу. Это может быть, например, специфичность, точность, правильность, верность, повторяемость, воспроизводимость, восстановление, способность обнаружения и прочность;
показатель функционирования – требования к рабочим характеристикам, согласно которым может быть оценен аналитический метод как адекватный для преследуемой цели и дающий надежные результаты;
допустимый уровень – максимальный уровень остатков, максимальный уровень или другое максимально допустимое отклонение для веществ, установленных в других национальных нормативных документах;
точность – степень приближения результатов независимых исследований, полученных в ранее установленных условиях. Мера точности обычно выражена в терминах неточности и вычисляется от стандартного отклонения результатов. Чем стандартное отклонение больше, тем меньше точность;
тест на опытность – анализ той же пробы, что позволяет лабораториям выбрать собственные методы исследования, при условии их использования в повседневной работе. Тест должен проводится в соответствии с международными инструкциями (ISO 43-1 и ISO 43-2) и может использоваться при оценке воспроизводимости методов;
качественный метод – метод анализа, идентифицирующий вещество на основе химических, биологических или физических свойств;
количественный метод – метод анализа, определяющий количество или массовую долю вещества таким образом, чтобы можно было выражать как численное значение соответствующих единиц;
тест-бланк реагента – полная процедура испытаний, применяемая при отсутствии испытуемой фракции или с использованием эквивалентного количества подходящего растворителя вместо исследуемой фракции;
восстановление – процент истинной концентрации вещества, возмещенного в течение аналитической процедуры. Это определяется во время признания (валидации) метода, при отсутствии сертифицированного референтного материала;
повторяемость – точность в условиях повторяемости;
условия повторяемости – условия, при которых результаты независимых испытаний получены тем же методом, на идентичных испытательных объектах, в той же лаборатории, тем же оператором, на том же оборудовании;
воспроизводимость – точность в условиях воспроизво¬димости;
условия воспроизводимости – условия, при которых результаты независимых испытаний получены тем же методом на идентичных испытательных объектах, в разных лабораториях, разными операторами, использующими разное оборудование;
прочность – чувствительность метода испытаний при изменении условий экспериментов, которые могут быть выражены как список типовых проб, аналитов, условий хранения, условий окружающей среды и/или подготовки пробы, при которых метод может применяться без или с незначительными изменениями. Для всех условий эксперимента, которые на практике могут варьировать (например, устойчивость реагентов, состав пробы, pH, температура), указываются все изменения, которые могут влиять на результат анализа;
тест-бланк пробы – полная процедура испытаний, применяемая к исследуемой части, отобранной из пробы, в которой аналит отсутствует;
метод выявления – метод, который используется для обнаружения наличия вещества или группы веществ в концентрациях, на определенном уровне. Эти методы имеют высокую производительную способность и используются для просеивания большого числа проб с целью выявления потенциально ложных результатов. Они разработаны, в частности, для исключения ложно соответствующих результатов;
внутрилабораторное изучение (внутреннее признание) – аналитическое изучение с привлечением одной отдельно взятой лаборатории, которая пользуется одним единственным методом анализа идентичных или различных испытуемых материалов, в разных условиях, оправданной длительности интервалов времени;
специфичность – способность метода различить измеряемый аналит от других веществ. Эта характеристика является преимущественно функцией описанной техники измерений, но может меняться в зависимости от группы веществ или матрицы;
стандартное дополнение – метод, при котором исследуемая проба разделена на две испытуемые фракции (или более). Одна часть собственно подлежит исследованию, а к остальным перед анализом добавляется известное количество стандарта аналита, определенное в пробе. Количество добавленного стандарта аналита должно располагаться между двукратным и пятикратным количеством аналита, определенного в пробе. Этот метод позволяет определять содержание аналита в пробе, учитывая собственное восстановление соответствующей процедуры испытаний;
эталон аналита – аналит известного и удостоверенного содержания и чистоты, служащий в качестве референтного при проведении анализа;
вещество – материал со специальным или определенным химическим строением и его метаболиты;
испытуемая фракция – количество материала, отобранного из пробы для испытаний, которое служит объектом анализа или исследования;
исследуемая проба – проба, приготовленная из лабораторной пробы, из которой должны быть отобраны испытуемые фракции;
правильность – степень приближения между средним значением, полученным из большого ряда результатов испытаний и принятой референтной величиной. Правильность обычно выражается как системная ошибка;
единица измерения – частная величина, определенная и принятая конвенцией, с которой сравниваются другие величины такого же рода, как результат измерений, в целях выражения их значения в сравнении с той величиной;
признание (валидация) – подтверждение экспертизой и обеспечение эффективных доказательств, что выполнены специальные требования для конкретного использования;
внутрилабораторая воспроизводимость – точность, полученная в той же лаборатории в установленных (предопределенных) условиях (относительно, например, метода, материалов для исследования, пользователей, окружающей среды) по оправданной длительности интервалов времени;
средство измерения – величина, измерительный аппарат, преобразователь, прибор, снаряжение, измерительная система, установка, а также референтный материал, используемый отдельно или совместно с одним или несколькими дополнительными приборами, поставляющими измерительную информацию.
Оборудование и средства измерения, используемые в методах или комбинациях методов испытаний, должны быть адекватными, узаконенными и метрологически поверенными в соответствии с требованиями Национальной метрологической системы.
Пробы должны быть получены, манипулированны и обработаны таким образом, чтобы возможности обнаружения вещества были максимальными. Типовые процедуры обращения с пробами должны предотвратить возможность случайного загрязнения или потери аналитов.
2.1.2. Выполнение испытаний
2.1.2.1. Восстановление
Во время анализа проб восстановление должно быть определено для каждой партии проб, если используется установленный коэффициент исправления восстановления. Если восстановление находится в рамках пределов, можно использовать установленный фактор коррекции. В противном случае необходимо использовать фактор восстановления, полученный для данной партии, за исключением случая, когда используется специальный фактор восстановления аналита в пробе – случай, когда необходимо использовать метод эталонных дополнений (пункт 3.5) или внутренний эталон для количественного определения аналита в пробе.
2.1.2.2. Специфичность
Метод должен обладать способностью различать аналит от других веществ в экспериментальных условиях. Следует представить оценку этой способности. Необходимо применять стратегии в целях предотвращения любого предвиденного совпадения с другими веществами, когда используется измерительная техника (например омологи, аналоги, метаболиты данного остаточного вещества). Очень важно анализировать любое совпадение, которое может быть спровоцированным составными элементами матрицы.
осадков и загрязнителей
Нижеперечисленные методы или комбинации методов рассматриваются как подходящие для идентификации органических остатков или загрязнителей для указанных групп веществ:
для органических остатков и загрязнителей
|
Способ измерения |
Вещество
|
Ограничения
|
|
1
|
2
|
3
|
|
Жидкостная хроматография (LC) или газовая хроматография (GC) с масс-спектрометрией |
Группы A и B
|
Только после поточного или автономного хроматографическогого разделения
Только в случае использования метода с полным просмотром или использования не менее 3-х (группа B) или 4-х (группа A) точек идентификации для методов, которые не регистрируют полный массовый спектр |
|
LC или GC с IR спектрометрическим обнаружением
|
Группы A и B
|
Должны соблюдаться специальные требования к инфракрасной (IR) спектрометрической абсорбции |
|
Тонкослойная хроматогра-фия с полным просмотром (DAD – обнаружение при помощи сети диодов) |
Группа B
|
Должны соблюдаться специальные требования к ультрафиолетовой (UV) спектрометрической абсорбции |
|
Флюоресценция LC
|
Группа B
|
Только для молекул с естественной флюоресценции и флюоресцирующих молекул после преобразования или расщепления на производные |
|
Тонкослойная хроматография с 2-D с полным просмотром UV/VIS
|
Группа B
|
Обязательны тонкослойная хроматография высокой чувствительности (HPTLC) двухмерной (2D) и co-хроматография |
|
Газовая хроматография (GC) – обнаружение с захватом электронов
|
Группа B
|
Только в случае использования двух колонок с разной полярностью
|
|
Иммунограмма LC
|
Группа B
|
Только в случае использования не менее двух разных хроматографических систем или второго независимого метода обнаружения |
|
LC-UV/VIS (единая длина волны)
|
Группа B
|
Только в случае использования не менее двух разных хроматографических систем или второго независимого метода обнаружения |
Методы подтверждения должны поставлять информацию о химической структуре аналита. В случае, когда ряд составных элементов дает ту же реакцию, метод не позволяет различать эти составные. Методы, основанные только на хроматографическом анализе, не предусматривающие использование спектрометрического обнаружения, не достаточны для использования в качестве методов подтверждения.
Если в методе используется внутренний эталон, подходящий к испытуемой фракции, он добавляется в начале экстракции. В зависимости от возможностей можно использовать устойчивые, помеченные изотопом формы аналита, рекомендованные в основном для масс-спектрометрического обнаружения, или вещества, структурно родственные с аналитом.
При невозможности использования подходящего внутреннего эталона, идентификация аналита подтверждается методом комбинировнной хроматографии. В этом случае должен быть получен только один пик, при этом пиковая высота или область идентификации пика является эквивалентной количеству добавленного аналита. При газовой хроматографии (GC) или жидкостной хроматографии (LC) ширина пика на половине максимальной высоты должна быть в диапазоне 90-110% первоначальной ширины, а задержки времени должны быть идентичными в пределах 5%. Для тонкослойной хроматографии (TLC) усиливается только предполагаемое пятно аналита; не появляются дополнительные пятна и не должен изменятся внешний вид.
Референтный или обогащенный материал, содержащий известные количества аналита в пределах или близко к разрешенному пределу или пределу обнаружения (несоответствующая контрольная выборка), так же как и соответствующие материалы контроля и образцы реагентов, предпочтительнее исследовать одновременно с каждой партией исследуемых проб, выполняя полную процедуру. Нагнетание экстрактов в аналитический инструмент осуществляется в следующем порядке: реагент рабочий, соответствующая контрольная проба, проба для подтверждения, новая соответствующая контрольная проба и, в завершение, несоответствующая контрольная проба. Любое изменение этой последовательности должно быть оправдано.
2.3.2. Дополнительные критерии эффективности функционирования и другие требования для количественных методов испытаний
2.3.2.1. Точность количественных методов
При повторных исследованиях сертифицированного референтного материала отклонение средней массовой фракции, коррелированной c экспериментально установленным восстановлением от сертифицированного значения, устанавливается в следующих пределах:
количественных методов
|
Массовая фракция |
Диапазон значения |
|
≤ 1 μг/кг
> 1 μг/кг до 10 μг/кг ≥ 10 μг/кг
|
–50 % до + 20 %
–30 % до + 10 % –20 % до + 10 % |
2.3.2.2. Точность количественных методов
Межлабораторный коэффициент вариации (CV) для повторного анализа референтного или обогащенного материала в условиях воспроизводимости не должен превышать уровень, подсчитанный при помощи уравнения Horwitz:
CV = 2(1-0,5logC),
где: C - массовая фракция, выраженная как мощность в 10-й степени (например, 1 миллиграмм/грамм = 10-3).
Примеры приведены в таблице 3.
в диапазоне массовых фракций аналита
|
Массовая фракция
|
Воспроизводимость CV (%)
|
|
1 μг/кг
10 μг/кг
100 μг/кг
1000 μг/кг (1 мг/кг)
|
(*)
(*)
23
16
|
|
(*) Для массовых фракций ниже 100 µг/кг применение уравнения Horwitz дает неприемлемые высокие значения. Следовательно, CV для концентраций ниже 100 µг/кг должен быть как можно ниже. |
|
Для испытаний, проведенных в условиях повторяемости, внутрилабораторный CV устанавливается обычно между 0,5 и 2/3 вышеуказанных значений. Для испытаний, проведенных в условиях внутрилабораторной повторяемости, внутрилабораторный CV не должен превышать воспроизводимость CV.
Для веществ, для которых установлен разрешенный предел метод должен достичь внутрилабораторной воспроизводимости не большей, чем значение воспроизводимости CV, соответствующей концентрации, равной 0,5 х разрешенный предел.
2.3.3. Критерии эффективности функционирования и другие требования, применяемые для обнаружения путем масс-спектрометрии
Методы масс-спектрометрии могут считаться подтверждающими методами только после линейного или автоматического хроматографического разделения.
2.3.3.1. Хроматографическое разделение
Для GC-MS – процедур газовое хроматографическое разделение проводится при помощи капиллярных колонок. Для LC-MS – процедур хроматографическое разделение должно проводиться при помощи приспособленных LC колонок. Во всех случаях минимально допустимое время задержки для исследуемого аналита превышает в два раза время задержки, соответствующее объему пустой колонки. Время задержки (или время относительной величины удерживания) аналита в испытуемой фракции будет соответствовать времени задержки эталона в пределах указанного интервала времени задержки. Интервал времени задержки пропорционален разрешающей способности хроматографической системы. Cоотношение между временем хроматографической задержки аналита и внутреннего стандарта, то есть времени относительной величины удерживания аналита, должно соответствовать таковому раствора калибровки при допуске толерантности ±0,5% для GC и ± 2,5 % для LC.
2.3.3.2. Масс-cпектрометрическое обнаружение
Масс-cпектрометрическое обнаружение проводится с использованием методов MS, таких как регистрация спектров полной массы (полный просмотр) или мониторинг избранных ионов (SIM - Selected Ion Monitoring), таких как мониторинг избранных реакций (Selected Reaction Monitoring), а также методов MS или MS - MSn, приспособленных, в комбинации с соответствующими способами ионизации. В массовой спектрометрии с высоким разрешением (HRMS) характерное разрешение должно быть больше 10 000 для полной массы с отклонением в пределах 10%.
Полный просмотр (сканирование): Если масс-спектрометрическое определение выполняется с регистрацией полных спектров, обязательно присутствие всех измеряемых диагностических ионов (молекулярный ион, аддукты, характерные для молекулярного иона, характерные фрагментированные ионы и ионы-изотопы) с относительной интенсивностью (напряженностью) более 10% в референтном спектре эталона.
SIM: в случае, когда масс-спектрометрическое определение выполняется методом фрагментографии, молекулярный ион является предпочтительным среди избранных диагностических ионов (молекулярные ионы, аддукты, характерные для молекулярного иона, характерные фрагментированные ионы и их изотопы). Избранные диагностические ионы не должны происходить исключительно из той же части молекулы. Отношение сигнал/шум для каждого диагностического иона должно быть ≥ 3:1.
Полный просмотр и SIM: относительные интенсивности обнаруженных ионов, выраженных в процентах от интенсивности (напряженности) наиболее интенсивного иона или интенсивного перемещения, должны соответствовать интенсивности эталона калибровки, либо растворов эталона калибровки или обогащенных проб при сравнимых концентрациях, измеряемых в тех же условиях, в пределах следующих допустимых отклонений:
интенсивности в различных методах масс-спектрометрии
|
Относительная интенсивность (% от основного пика) |
EI-GC-MS (относительный) |
CI-GC-MS, GC-MSn LC-MS, LC-MSn (относительные) |
|
> 50 %
> 20 % до 50 %
> 10 % до 20 %
≤ 10 %
|
±10 %
±15 % ±20 % ±50 % |
±20 %
±25 % ±30 % ±50 % |
Интерпретация массовых спектральных данных: относительные интенсивности диагностических ионов и/или пары иона – предшественника/продукта идентифицируются путем сравнения спектров или объединения сигналов каждого выделенного массового следа. В случае применения основного исправления оно равномерно на всей партии (см. 2.3.1, абзац четвертый) и четко обозначено.
Полный просмотр: в случае, когда полные спектры просмотра зарегистрированы в простой массовой спектрометрии, минимум четыре иона должны присутствовать при относительной интенсивности (напряженности) ≥ 10% от основного пика. Молекулярный ион должен быть включен в случае, когда он присутствует в референтном спектре с относительной интенсивностью (напряженностью) ≥ 10 %. По меньшей мере, четыре иона должны находиться в максимально допустимых пределах для относительной интенсивности иона (таблица № 5). Можно использовать автоматизированный библиографический поиск. В этом случае сравнение массовых спектральных данных в исследуемых пробах с данными растворов калибровки должно превысить критический фактор сравнения. Этот фактор определяется в течение процесса признания (валидации) для каждого аналита на основе спектров, для которых выполнены нижеописанные критерии. Проверяется изменчивость спектров, вызванных типовой матрицей и применением детектора.
СИМ: Если измеряются массовые фрагменты при помощи других методов, кроме полного просмотра, используется бальная (пункты) система идентификации для интерпретации данных. Для подтверждения веществ с анаболическим эффектом и неавторизованных веществ, стильбенов, их производных, солей и эфиров, антитироидов, стероидов, лактонов резорциновой кислоты, в том числе зереанол, бета-агонистов (в дальнейшем – вещества группы А), требуется не менее 4 пунктов. Для подтверждения медикаментов ветеринарного пользования и других загрязнителей для продуктов животного происхождения (противомикробные вещества, включая сульфаниламиды и хинолоны; другие медикаменты ветеринарного пользования, в том числе антигельминтики, кокцидиостатики, включая нитронидазол, карбаматы и перитроиды, транквилизаторы, противовоспалительные нестероиды, другие фармакологически активные вещества; другие вещества и загрязнители среды, хлорорганические соединения, фосфорорганические соединения, химические элементы, микотоксины, красители, другие вещества, в том числе другие неавторизованные вещества ветеринарного назначения (в дальнейшем – вещества группы B) необходимо не менее 3 пунктов идентификации. В таблице 5 указано число пунктов идентификации, которое может получить каждый из основных методов масс-спектрометрии. Несмотря на это, для того, чтобы выполнить условия по пунктам идентификации, необходимым для подтверждения и для определения суммы пунктов идентификации:
a) измеряется, по крайней мере, одно отношение иона;
b) все измеряемые релевантные отношения иона должны отвечать вышеописанным критериям;
c) могут быть объединены не более трех отдельных методов для достижения минимального количества пунктов идентификации.
и полученными пунктами идентификации
|
Методика MS
|
Полученные пункты идентификации на ион |
|
Масс-спектрометрия низкого разрешения (LR) LR-MSn иона предшественника
LR-MSn 1,5 продуктов перемещения HRMS
HR-MSn иона-предшественника
HR-MSn продуктов перемещения |
1,0
1,0
1,5
2,0
2,0
2,5
|
Примечания:
1. Каждый ион может быть подсчитан только один раз.
2. GC-MS ионизация путем электронной бомбардировки расценена как отличающаяся от методики GC-MS с химической ионизацией.
3. Для увеличения числа пунктов идентификации можно использовать больше аналитов только в том случае, когда производные проявляют различные химические реакции.
4. Для веществ группы А, если используется один из следующих методов в аналитической процедуре: HPLC вместе с диодной матрицой с полным просмотром фотоспекирометрии (DAD); HPLC вместе с обнаружением путем флюоресценции; HPLC, соединенный к иммунограммой; двумерный TLC вместе с спектрометрическим обнаружением; это позволит повысить не более чем на один пункт идентификации, при условии выполнения критериев, релевантных для этих методов.
5. Промежуточные продукты включают и продукты второго и третьего поколения.
методов и комбинаций методов (n = целое число)
|
Метод (методы)
|
Количество ионов
|
Пункты идентификации
|
|
1
|
2
|
3
|
|
GC-MS (EI или CI) |
N
|
n
|
|
GC-MS (EI и CI) |
2 (EI) + 2 (CI) |
4
|
|
GC-MS (EI или CI) 2 производные |
2 (производное A) + 2 (производное B) |
4
|
|
LC-MS
|
N
|
n
|
|
GC-MS-MS
|
1 ион - предшественник и 2 из второго поколения |
4
|
|
LC-MS-MS
|
1 ион - предшественник и 2 из второго поколения |
4
|
|
GC-MS-MS
|
2 иона - предшественника, по одному иону из второго поколения |
5
|
|
LC-MS-MS
|
2 иона - предшественника, по одному иону из второго поколения |
5
|
|
LC-MS-MS-MS
|
1 ион - предшественник, один ион из второго поколения и один ион из третьего поколения |
5,5
|
|
HRMS
|
N
|
2n
|
|
GC-MS şi LC-MS |
2 + 2
|
4
|
|
GC-MS şi HRMS |
2 + 1
|
4
|
2.3.4. Критерии эффективности функционирования и другие требования для хроматографии c инфракрасными лучами
Адекватные пики: адекватные пики представляют максимальное поглощение в инфракрасном спектре эталона калибровки, выполненное в соответствии с приведенными ниже требованиями.
2.3.4.1. Инфракрасное обнаружение
Максимальное поглощение должно быть в диапазоне волн 4000-500 см-1.
Интенсивность (напряженность) поглощения не должна быть меньше:
a) специфичной молярной абсорбции равной 40 относительно пиковой базовой линии; или
b) относительной абсорбции равной 12,5% от абсорбции наиболее интенсивного пика в области 4000-500 см-1.
В случае, когда эти две абсорбции измеряются относительно нулевой абсорбции и при 5% от абсорбции наиболее интенсивного пика в диапазоне 4000-500 см-1, тогда обе две абсорбции измеряются относительно базовой линии пика.
Примечания: Следует отметить, что хотя с теоретической точки зрения предпочтительнее пики, измеренные в соответствии с подпунктом a), на практике пики проще определять согласно подпункту b).
Число пиков в инфракрасном спектре аналита чьи частоты соответствуют адекватному пику спектра эталона калибровки, с отклонением ± 1 см-1 .
2.3.4.2. Интерпретация инфракрасных спектральных данных
Поглощение должно присутствовать во всех областях спектра аналита, которые соответствуют адекватному пику спектра референтного эталона калибровки. Требуется не менее шести адекватных пиков в инфракрасном излучении спектра эталона калибровки. Если имеется менее шести адекватных пиков, данный инфракрасный спектр не может использоваться в качестве референтного спектра. «Результат», то есть процент адекватных пиков аналита в инфракрасном спектре, должен быть не менее 50. В случае, когда нет точных данных для адекватного пика, соответствующая область спектра аналита должна соответствовать присутствию пика с тем же уровнем. Процедура применяется только к пикам поглощения спектра пробы с интенсивностью (напряженностью) не менее трехкратной высоты фонового шума.
2.3.5. Критерии эффективности функционирования и другие требования для определения аналита путем LC совместно с другими методами обнаружения
2.3.5.1. Хроматографическое разделение
Используется внутренний эталон в случае, если для этой цели доступен подходящий материал. Предпочтительнее родственный эталон с временем задержки, близкой к времени задержки аналита. Аналит будет элюировать во времени задержки, типичной для соответствующего эталона калибровки, в тех же экспериментальных условиях. Минимально допустимым временем задержки для аналита должно быть двухкратное время задержки, соответствующее объему пустой колонки. Отношение между временем задержки аналита и временем задержки внутреннего эталона, то есть время относительного удерживания аналита, должно быть равным времени эталона в соответствующей матрице, с отклонением ± 2,5%.
2.3.5.2. Полный просмотр с UV/VIS обнаружением (ультрафиолетовое/видимый свет)
Необходимо выполнить критерии эффективности функционирования, применеяемые к LC-методам.
Максимальное поглощение в спектре аналита должно представлять ту же длину волны, что и эталон, в пределах, определенных разрешающей способностью системы обнаружения. Для обнаружения при помощи диодной матрицы это значение обычно находится в промежутке
± 2 nm. Для частей двух спектров, относительная абсорбция которых равна или более 10%, спектр аналита более 220 nm не должен отличаться от спектра эталона калибровки. Этот критерий выполняется в случаях когда, во-первых, присутствуют те же максимальные пределы, и, во-вторых, когда разность между двумя спектрами ни в одной точке не наблюдалась больше 10% абсорбции эталона калибровки. Если проводятся исследования и сравнение с базой данных, разница сравнения спектральных данных испытуемых проб с таковыми раствора калибровки должна превысить критический фактор сравнения. Этот фактор должен определяться в течение процесса валидации (признания) для каждого аналита на основе спектров, для которых выполнены вышеописанные критерии. Проверяется изменчивость спектров, вызванная типовой матрицей и применением детектора.
2.3.5.3. Критерии эффективности функционирования для флюорометрического обнаружения
Должны соблюдаться критерии эффективности функционирования, применяемые к LC-методам.
Это применяется к молекулам с естественной флюоресценцией и к молекулам, которые показывают флюоресценцию после любого преобразования или расщепления. Выбор длины волн возбуждения и эмиссии в комбинации с хроматографическими условиями должен осуществляться таким образом, чтобы минимизировать появление интерферирующих компонентов в бланковых экстрактах.
Максимальная величина самого близкого пика в хроматограмме должна отделяться от обозначенного пика аналита, по меньшей мере, на измеренную полную ширину пика на 10% максимальной высоты пика аналита.
2.3.5.4. Критерии эффективности функционирования для определения аналита путем иммунограммы LC
Иммунограмма LC как таковая не подходит для использования в качестве подтверждающего метода.
Необходимо выполнить критерии функционирования, применяемые к LC-методам.
Предопределенные параметры контроля качества, например, неспецифическая фиксация или относительная фиксация контрольных проб или значение абсорбции бланка должны быть в пределах, полученных в течение валидации (пригодности) анализа.
Иммунограмма составляется, по меньшей мере, из пяти фракций.
Каждая фракция должна быть меньше, чем половина ширины пика.
Фракция с максимальным содержанием аналита должна быть идентичной для подозреваемой пробы, не соответствующей контрольной пробе и эталону.
2.3.5.5. Определение аналита путем LC с UV/VIS – обнаружением (единая длина волны)
LC с UV/VIS – обнаружением (единая длина волны) не подходит для использования в качестве подтверждающего метода.
Максимальная величина самого близкого пика в хроматограмме должна отделяться от обозначенного пика аналита, по меньшей мере, на измеренную полную ширину пика на 10% от максимальной высоты пика аналита.
2.3.6. Критерии эффективности функционирования и другие требования, которые применяются для определения аналита путем 2-D TLC совместно с UV/VIS – спектрометрическим обнаружением с полным просмотром (сканированием)
Обязательны двумерная HPTLC и комбинированная хроматография.
Значение RF аналита должно соответствовать значениям RF эталонов с отклонением ±5%.
Внешний вид аналита не должен отличаться от такового эталона.
Для пятен того же окрашивания центр самого близкого пятна должен быть отделен от центра пятна аналита на расстояние, равное, по меньшей мере, половине суммы диаметров световых пятен.
Спектр аналита визуально не должен отличаться от спектра стандарта, в соответствии с описанием для полного сканирования (просмотра) UV/VIS – обнаружением.
Если проводятся исследования и сравнение с базой данных, разница сравнения спектральных данных испытуемых проб с таковыми раствора калибровки должна превышать критический фактор сравнения. Этот фактор должен определяться в течение процесса валидации (признания) для каждого аналита на основе спектров, для которых выполнены вышеописанные критерии. Проверяется изменчивость спектров, вызванное типовой матрицей и применением детектора.
2.3.7. Критерии функционирования и другие требования, которые используются при определении аналита с применением GC – обнаружения путем захвата электронов
Используется внутренний эталон, если для этой цели доступен подходящий материал. Предпочтительнее родственное вещество с временем задержки, близким к времени задержки аналита. Аналит должен элудировать во время задержки, типичное для соответствующего эталона калибровки в тех же экспериментальных условиях. Минимально допустимое время задержки для аналита должно быть двухкратным времени задержки, соответствующему объему пустой колонки. Соотношение между временем задержки аналита и временем задержки внутреннего эталона, то есть временем относительного удерживания аналита, должно быть равным времени задержки эталона калибровки в соответствующей матрице с отклонением ± 0,5%.
Максимальная величина самого близкого пика в хроматограмме должна отделяться от пика аналита не менее чем на полную ширину пика, измеренную при 10% от максимальной высоты пика аналита. Для дополнительной информации можно использовать комбинированную хроматографию.
Следующие методы или комбинации методов рассматриваются подходящими для идентификации химических элементов, включенных в таблицу 7.
методы для химических элементов
|
Методика
|
параметр
|
|
Анодная вольтметрия с дифференциальным импульсом (DPASV)
Атомно-абсорбционная спектрометрия (AAS) Пламенная фотометрия Производство гидритов Холодный пар Электротермическая атомизация (графитовая печь) Спектрометрия атомной эмиссии (AES) Плазменная индукция Масс-спектрометрия (MS) Плазменная индукция |
Электрический сигнал
Длина волны поглощения Длина волны поглощения Длина волны поглощения Длина волны поглощения Длина волны эмиссии Соотношение масса/заряд (m/z) |
2.4.1. Общие критерии эффективности функционирования и другие требования, применяемые к подтверждающим методам
Референтный или обогащенный материал, содержащий известные количества аналита, уровня близкого или максимально разрешенного или предела решения (несоответствующая контрольная проба), а также соответствующие материалы контроля и необработанные бланки реагента, предпочтительно исследовать одновременно с каждой партией проанализированных проб с использованием полного метода. Рекомендуется нагнетание экстрактов в аналитический инструмент в следующем порядке: бланк-реагент, соответствующая контрольная проба, проба для подтверждения, другая соответствующая контрольная проба и, в итоге, несоответствующая контрольная проба. Любое другое изменение порядка должно быть оправдано.
Как правило, большая часть аналитических методов требует полного вываривания органической матрицы, чтобы получить растворы до определения аналита. Это может быть достигнуто при помощи процедур минерализации микроволнами, которые сводят до минимума риск потери и/или загрязнения данных аналитов. Используются дезактивированные тефлоновые сосуды хорошего качества. В случае, когда используются другие методы влажного или сухого вываривания, должны существовать идентифицируемые пробы, позволяющие исключить возможные явления потери или загрязнения. Вместо вываривания, процедуры разделения (например извлечение) могут, в некоторых условиях, быть выбраны для отделения аналита от матричных компонентов и/или для концентрирования аналита в целях его введения в аналитическое оборудование.
Что касается калибровки, внешней или основанной на эталонном методе дополнения, рекомендуется принимать меры по предотвращению превышения рабочей зоны, установленной для анализа. В случае внешней калибровки обязательно, чтобы эталоны калибровки были подготовлены из раствора, состав которого наиболее приближен к составу раствора пробы. Фоновое исправление также должно применяться в случае, если этого требуют специфические условия испытаний.
2.4.2. Критерии функционирования и другие дополнительные требования, которые применяются к количественным методам
2.4.2.1. Правильность количественных методов
При повторных исследованиях сертифицированного референтного материала (CRM) для химических элементов экспериментально определенное отклонение среднего содержания от сертифицированного значения должно находиться в пределах ±10%. В случае, когда не доступен ни один CRM такого типа, допустимо, чтобы правильность измерений оценивалась путем восстановления дополнений известных количеств элементов к неизвестным пробам. Следует отметить, что в отличие от аналита, добавленный элемент химически не связан в реальной матрице и, следовательно, полученные этим методом результаты имеют меньшее признание, чем полученные с помощью CRM. Данные восстановления допустимы только в случае их нахождения в пределах ±10% от значения мишени.
2.4.2.2. Точность количественных методов
В случае повторного анализа пробы, проведенного в условиях внутрилабораторной воспроизводимости, внутрилабораторный коэффициент вариации (CV) середины не должен превышать значений, приведенных в таб-
лице 8.
в диапазоне массовых фракций элемента
|
Массовые фракции |
CV (%)
|
|
≥ 10 μг/кг до 100 μг/кг
> 100 μг/кг до 1000 μг/кг ≥ 1000 μг/кг |
20
15
10
|
2.4.3. Специфические требования, применяемые при анодной вольтметрии с дифференциальным импульсом (DPASV)
Полное разрушение органического вещества в пробах до DPASV определения имеет самое большое значение. В вольтамограммах не должен быть замечен ни один широкий сигнал, исходящий из присутствия органических материалов. Следовательно, квантификация должна быть выполнена методом эталонных дополнений. Одновременно с методом должны быть поставлены образцы типичных вольтамограмм типового раствора.
2.4.4. Специальные требования, применяемые к атомно-абсорбционной спектрометрии (AAS)
Этот метод является по сути методом моноэлементным и, следовательно, требует оптимизации экспериментальных условий в зависимости от элемента, который будет определен количественно. По возможности, результаты должны быть предметом качественной и количественной проверки, прибегая к альтернативным линиям поглощения (в идеале отбираются две различные линии абсорбции). Эталоны готовятся из жидкой матрицы, максимально приближенной к исследуемой жидкости (например, в концентрациях кислоты или состава измененного агента). Чтобы минимизировать незаполненные значения, все реагенты должны быть самой высокой доступной чистоты. В зависимости от выбранного способа для испарения и/или распыления пробы могут быть различные типы AAS.
2.4.4.1. Специфические требования, применяемые к пламенной ААС
Регулировка инструментов проводится для каждого элемента. В частности, необходимо контролировать состав и дебит газа. Во избежание взаимодействия из-за абсорбции фона используется корректор с постоянным источником. В случае неизвестных матриц, проверяется необходимость фоновой коррекции.
2.4.4.2. Специфические требования, применяемые для ААС с графитной печью
Лабораторное заражение часто влияет на точность, особенно когда ведется работа на уровне ультраследов в графитовой печи. Следовательно, необходимо использовать реактивы повышенной чистоты, деминерализированную воду и инертный пластиковый материал для манипуляции проб и эталонов. Необходима оптимальная регулировка инструментов для каждого элемента. В частности, проверяются условия предобработки и атомизации (температура, время) и изменение матрицы.
Работа в условиях изотермической атомизации (например, графитная колонка с поперечным утеплением со встроенной платформой Львова) уменьшает влияние матрицы относительно атомизации аналита. Изменение матрицы, комбинированное с фоновой коррекцией Зеемана, позволяет квантификацию с помощью стандартной кривой, основанной на измерениях стандартных водных растворов.
2.4.5. Специфические требования, применяемые к атомно-абсорбционной спектрометрии с образованием гидратов
Органические соединения, содержащие такие элементы как мышьяк, висмут, германий, свинец, антимоний, селен, станий и теллур, могут быть очень устойчивы и требуют окислительного разложения для получения правильных результатов относительно полного содержания элементов. Следовательно, рекомендуется пищеварительный метод с помощью микроволн или кальцинирование при высоком давлении в условиях сильного окисления. Необходимо уделить наибольшее внимание полной и воспроизводительной конверсии элементов в соответствующих гидритах.
Образование гидрида мышьяка в растворе соляной кислоты с NaBH4 зависит от состояния окисления мышьяка (Аs III: быстрое образование, As V: медленное образование). Во избежание потери чувствительности при определении As V с использованием техники поточной инъекции, спровоцированной коротким временем реакции данной системы, As V должен быть доведен до As III после разложения путем окисления. Йодид калия/аскорбиновая кислота или цистеин адекватны для данной цели. Бланки, стандартные растворы и пробные растворы обрабатываются тем же способом. Система по партиям позволяет определить два типа мышьяка, не влияя на точность. Из-за более длинного периода образования гидрида Аs V калибровка производится путем интеграции поверхностей пиков. Необходимо оптимизировать регулировку инструментов. Важно контролировать поток газа, переводящий гидрит к атомизатору.
2.4.6. Специфические требования, применяемые к атомно-адсорбционной спектрометрии в фазе холодного пара
Холодный пар используется только для ртути. В случае потери начальной ртути при испарении и адсорбции необходимо особое внимание на всем протяжении тестирования. Следует осторожно избегать загрязнения через среды и реактивы.
В случае органических соединений, содержащих ртуть, необходим распад путем окисления для получения правильных результатов, относящихся к общему содержанию ртути. Для распада рекомендуется использовать опечатанные системы распада микроволнами или кальцинацией под высоким давлением. При очистке оборудования, входящего в контакт со ртутью, требуется особое внимание.
Техника поточной инъекции предоставляет преимущества. Для минимальных уровней решения рекомендуется адсорбция первоначальной ртути абсорбентом и/или платиной с последующей термической десорбцией. Контакт абсорбента или клетки с влажностью нарушает измерение ртути и его следует избегать.
2.4.7. Специфические требования, применяемые к атомно-эмиссионной спектрометрии, соединенной с индуктивной плазмой (ICP-AES)
Атомно-эмиссионная спектрометрия, соединенная с индуктивной плазмой (ICP-AES), является методом мультиэлементным, позволяющим одновременное измерение нескольких элементов. Для использования ICP-AES пробы должны быть заранее разморожены для расщепления органической матрицы. Используются опечатанные системы переваривания при помощи микроволн или кальцинирования под высоким давлением. Для того, чтобы анализ ICP-AES был эффективным, важно эталонирование инструмента и подбор элементов или длины волн. Для эталонирования прибора в случае линейных эталоных кривых обычно необходимо измерять растворы для эталонирования только по четырем концентрациям, поскольку в целом кривые для эталонирования ICP-AES являются линейными на от четырех до шести степенях величины концентрации. Калибровка системы ICP-AES, как правило, должна осуществляться с мультиэлементным эталоном, который должен быть подготовлен в растворе, содержащем ту же концентрацию кислоты, что и раствор для измерения. Для линейной кривой должна быть проверена концентрация элементов.
Выбор длины волн для оценки выбросов от аналитов должен быть адекватным для концентрации элементов, которые следует определить. Там, где концентрация аналита выходит из зоны действия линии эмиссии, используется другая линия эмиссии. Наиболее чувствительная линия эмиссии (без вмешательства) выбирается первой, за ней следует линия менее чувствительная. В случае, если используется на пределе обнаружения или в его близости, лучшим выбором, как правило, является линия эмиссии, наиболее чувствительная для соответствующего аналита. В ICP-AES основные трудности исходят от спектральных помех или фона. Возможными вмешательствами являются, например, несоответствие фонового шума, производная фонового шума, слабое спектральное решение и случайные вариации фонового шума. Каждая интерференция имеет свои собственные причины и способы их устранения. Применяется, в зависимости от матрицы, коррекция интерференции и оптимизация эксплуатационных параметров. Некоторые интерференции можно избежать путем разбавления или адаптации матриц. Для каждой исследуемой партии проб для тестирования образцы референтного материала и обогащенный материал, содержащие известные количества аналита и аналитов, а также бланк обрабатываются так же, как и проба для тестирования. Для контроля возможных отклонений эталон должен проверяться после 10 проб. Все реагенты и газ плазмы должны быть самой высокой возможной чистоты.
2.4.8. Особые требования, применяемые к масс-спектрометрии с плазменной индукцией (ICP-MS)
Определение следов элементов со средней атомной массой, таких как хром, медь и никель, может быть серьезно нарушено другими изобарными и многоатомными ионами. Этот феномен можно избежать только при наличии силы резолюции не менее 7000-8000. Трудностями, связанными с техникой MS, в частности, являются отклонения инструмента, эффекты матрицы и молекулярные ионные пертурбации (m/z < 80). Необходима внутренняя многоразовая калибровка, охватывающая те же массовые диапазоны, что и определяемые элементы для коррекции отклонения инструмента и эффектов матрицы.
Необходимо, чтобы полное разложение органических материалов в пробах производилось до измерения в ICP-MS. Как и в AAS, после распада в опечатанных контейнерах легко испаряющиеся элементы, например йод, должны быть переведены в состояние стабильного окисления. Наиболее важная интерференция исходит от молекулярных ионных соединений аргона (плазменный газ), водорода, углерода, азота и кислорода (кислоты распада, примесей плазмы и участвующих атмосферных газов) и матрицы пробы. Полный распад, измерения фона, надлежащий выбор проанализированных масс, иногда связанные с низким насыщением (низкий уровень выявления) и кислотами распада, на пример, азотной кислоты, необходимы для избежания интерференции.
Для определяемых элементов интерференции исключаются путем выбора соответствующих проанализированных масс, в том числе путем подтверждения отношений изотопов. Ответ инструмента проверяется с учетом фактора Fano для каждого измерения, используя внутренние эталоны.
3. Признание (валидация)
Валидация должна доказать, что метод испытаний отвечает требованиям, применяемым в случаях релевантных характеристик функционирования.
Проверки в разных целях требуют различных категорий методов. В таблице 9 приведены функциональные характеристики, которые проверяются по каждому типу метода.
характеристикам функционирования,
которые следует определить
|
|
Лимит обнаружения CCβ
|
Лимит решимости CCα
|
Точность/
исправление
|
Достоверность
|
Селективность/ специфичность
|
Применимость
достоверность/
стабильность
|
|
|
Качественные методы
|
S
|
+
|
–
|
–
|
–
|
+
|
+
|
|
C
|
+
|
+
|
–
|
–
|
+
|
+
|
|
|
Количественные методы
|
S
|
+
|
–
|
–
|
+
|
+
|
+
|
|
C
|
+
|
+
|
+
|
+
|
+
|
+
|
|
|
S = методы обнаружения; C = методы подтверждения; + = обязательное определение |
|||||||
Признание может также осуществляться путем использования межлабораторных исследований, приведенных в Codex Alimentarius, ISO или IUPAC, либо других методов, таких как внутрилабораторные исследования или внутреннее признание. Настоящая глава основана на внутрилабораторных исследованиях (или внутреннем признании) и носит модулярный подход. Такой подход состоит в:
1) гамме совместных функциональных характеристик, не зависимых от используемой модели признания;
2) специфических процедурах, связанных с моделью, в соответствии с описанием, приведенным в таблице 10.
зависимые и не зависимые от модели
|
Признание
|
||
|
Параметры функционирования, не зависимые от модели |
Параметры функционирования, зависимые от модели
|
|
|
Общие характеристики функционирования (3.1.1)
|
Классическое признание (3.1.2)
|
Внутреннее признание (3.1.3)
|
|
Специфичность
Точность
Устойчивость: минимальные отклонения Стабильность
|
Восстановление
Повторяемость
Воспроизводимость
внутрилабораторная
Повторяемость
Предел решимости (CCα)
Способность обнаружения (CCβ)
Стандартная кривая Устойчивость: значительные отклонения |
Восстановление
Повторяемость
Воспроизводимость
внутрилабораторная
Повторяемость
Предел решимости (CCα)
Способность обнаружения (CCβ)
Стандартная кривая Устойчивость
|
3.1.1. Рабочие характеристики, независимые от модели
Независимо от подхода, выбранного для признания, должны быть определены характеристики функционирования, указанные в дальнейшем. Для уменьшения рабочей нагрузки может использоваться хорошо задуманный и статистически признанный подход для совмещения полученного опыта по определению различных параметров.
3.1.1.1. Специфичность
Для методов испытаний важна способность различить аналит от родственных веществ (изомеров, метаболитов, продуктов распада, эндогенных веществ, составляющих матриц и т.д.) Необходимы два подхода для проверки наличия интерференций.
В результате потенциально интерферентные вещества изолируются и анализируются релевантные пробы-бланк для обнаружения присутствия возможных интерференций и оценки их эффектов:
выбирается набор химически родственных соединений (метаболиты, производные, и т.д.) или другие вещества, которые можно встретить в пробе одновременно с соответствующим продуктом;
анализируется соответствующее число представительных проб-бланк (n ≥ 20) и контролируется наличие интерференций (сигналов, пиков, следы ионов ) в данной области, в которой предполагается, что элюирует аналит-мишень;
также обогащаются представительные пробы-бланк веществами до соответствующих концентраций, которые могут интерферировать с идентификацией и/или квантификацией аналита;
после исследования наблюдается, если:
а) присутствие интерференций смогло привести к ложной идентификации;
b) присутствие одной или нескольких интерференций препятствует идентификации аналита-мишени;
с) квантификация существенно затронута.
3.1.1.2. Правильность
Настоящий пункт описывает определение правильности (являющейся одним из элементов точности). Правильность может быть установлена только при помощи сертифицированного общепринятого материала (CRM). Необходимо использовать CRM во всех случаях, если он доступен. Метод описан подробно в ISO 5725-4. Ниже приведен пример:
проводится 6-кратный анализ CRM в соответствии с инструкциями по тестированию, применяемыми для метода;
определяется концентрация присутствующего aнaлита в каждой последовательной пробе;
вычисляется среднее стандартное отклонение и коэффициент вариаций (%) для этих концентраций;
вычисляется правильность путем деления обнаруженной средней концентрации на достоверное значение (измеряемое в концентрации) и умножается на 100 для выражения результата в процентах.
Правильность (%) = средняя обнаруженная концентрация, исправленная с возмещением Ч 100/достоверное значение.
В отсутствие CRM можно определить возмещение правильности вместо точности.
3.1.1.3. Применяемость/прочность (незначительные вариации)
К такому методу испытания прибегают для введения незначительных разумных вариаций и наблюдения их последствий.
Предварительные исследования должны осуществляться выбором факторов перед обработкой, очищением и исследованием пробы, которые могут повлиять на результаты исследования. Такими факторами могут быть потребитель, источник и срок годности реактивов, растворители, эталоны и экстракты эталона, скорость обогревания, температура, pH и множество других факторов, которые могут появиться в лаборатории. Они могут быть изменены на один порядок соответствующего размера вариаций, встречаемых между разными лабораториями.
Идентифицируются возможные факторы, которые могут повлиять на результаты.
Легко варьируется каждый фактор.
Производится тест прочности по методу Youden (на данной стадии могут использоваться и другие допустимые методы. Тем не менее подход Youden сводит к минимуму необходимые время и усилия). Подход Youden является фракционно-факторным методом. Взаимодействия между разными факторами не являются выявляемыми.
В случае если один фактор существенно влияет на результаты измерений, проводятся дополнительные исследования, чтобы определить приемлемые пределы этого фактора.
Факторы, значительно влияющие на результаты, должны быть четко идентифицированы в протоколе исследования.
Основой не является изучение одного колебания, а одновременное введение больших вариаций. Например, отметим буквами A, B, C, D, E, F и G номинальные значения 7 разных факторов, которые могут повлиять на результаты исследования в случае, если их номинальные значения легко изменены. Укажем другие значения тех же факторов строчными буквами a, b, c, d, e, f и g. Получаем 27 или 128 разных потенциальных комбинаций.
Можно выбрать поднабор из 8 таких комбинаций, отображающих равновесие между прописными и строчными буквами (таблица 11). Осуществляются 8 определений с использованием по одной комбинации выбранных факторов (A-G). Результаты определения изображаются буквами S–Z из таблицы 11.
(незначительные вариации)
|
Значение фактора F |
Число исследуемых комбинаций |
|||||||
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
|
A/a
B/b
C/c
D/d
E/e
F/f
G/g
|
A
B
C
D
E
F
G
|
A
B
c
D
e
f
g
|
A
B
C
d
E
f
g
|
A
B
c
d
e
F
G
|
A
B
C
d
e
F
g
|
A
B
c
d
E
f
G
|
A
B
C
D
e
f
G
|
A
B
c
D
E
F
g
|
|
Наблюдаемый результат R
|
S
|
T
|
U
|
V
|
W
|
X
|
Y
|
Z
|
|
Для подсчета см. примеры исследований по прочности в пункте 3.3 |
||||||||
3.1.1.4. Стабильность
Обнаружено, что недостаточная стабильность аналита или составных частей матрицы в пробе на протяжении хранения или анализа может вызывать значительные отклонения уровня результата анализа. Необходимо также контролировать стабильность эталона в растворе. В общем стабильность аналита в разных условиях хранения хорошо известна. Контроль условий хранения является частью нормальной системы аккредитации лабораторий. В случае, если стабильность неизвестна, она может быть установлена в соответствии с приведенными в дальнейшем примерами.
Стабильность аналита в растворе:
Готовятся свежие основные растворы аналита (аналитов) и разбавляются в соответствии с инструкциями по исследованию для получения аликвотных частей в достаточном количестве (например, сорок) из каждой отобранной концентрации (близко к пределу минимального функционирования, необходимого для веществ, для которых не установлен ни один разрешенный предел, или близко к пределу, разрешенному для остальных веществ). Готовятся растворы аналита, используемые для обогащения и в конечном растворе, а также любой другой интересующий раствор (например, производные эталона).
Измеряется содержимое аналита в свежеприготовленном растворе в соответствии с инструкциями по исследованию.
Наливаются соответствующие объемы в приспособленные сосуды, этикетируются и ставятся на хранение согласно следующему плану:
стабильности аналита в растворе
|
|
–20 °C
|
+4 °C
|
+20 °C
|
|
Тень
|
10 аликвотных частей
|
10 аликвотных частей
|
10 аликвотных частей
|
|
Свет
|
10 аликвотных частей
|
10 аликвотных частей
|
10 аликвотных частей
|
Установленное время хранения составляет 1, 2, 3 и 4 недели или более, по необходимости, например, до момента появления первых феноменов распада во время идентификации и/или квантования. Максимальное время и оптимальные условия хранения регистрируются.
Определение концентрации аналита (аналитов) в каждой аликвотной части производится во время исследования с учетом концентрации свежеприготовленного 100% раствора аналита.
Остаточный аналит % = Ci Ч 100/Cсвежий
Ci = концентрация в учтенный момент;
Cсвежий = концентрация свежего раствора.
Стабильность аналита или аналитов в матрице
По мере возможности используются естественным образом зараженные пробы. В отсутствие естественным образом зараженного материала рекомендуется использовать матрицу, обогащенную аналитом.
В естественно зараженном материале определяется концентрация, пока он еще свежий. Отбираются и другие аликвотные части после 1, 2, 4 и 20 недель и определяются концентрации. Материал должен храниться при минимальной температуре -20°C или ниже, по необходимости.
При отсутствии естественным образом зараженного материала отбирается материал, не содержащий аналит, и гомогенизируется. Материал делится на пять аликвотных частей. В каждую аликвотную часть добавляется аналит, изготовленный предпочтительно, в небольшом количестве водного раствора. Сразу исследуется одна аликвотная часть. Остальные аликвотные части хранятся при температуре -20°C или ниже, по необходимости, и исследуются через 1, 2, 4 и 20 недель.
3.1.1.5. Градуировочные кривые
Если градуировочные кривые используется для квантификации:
для построения кривой используется не менее пяти уровней (включая нулевой);
описывается зона действия градуировочной кривой;
описывается математическая модель кривой и приспосабливаются данные к кривой;
описываются допустимые уровни параметров кривой.
В случае необходимости серийного эталонирования, основанного на стандартном растворе, допустимые диапазоны должны быть обозначены для параметров градуировочной кривой, которые могут варьировать от одной серии к другой.
3.1.2. Классические процедуры признания
Для вычисления параметров классическими методами необходимо провести множество исследований. Каждая функциональная характеристика должна быть определена для каждого важного изменения (см. предыдущие отметки по применяемости/прочности). При предварительном устранении возможных интерференций, многоаналитными методами одновременно может исследоваться несколько аналитов. Несколько функциональных характеристик может определяться в одном и том же порядке. Таким образом, для сокращения труда рекомендуется комбинировать, насколько возможно, исследования (например, повторяемость и внутрилабораторная воспроизводимость со специфичностью, анализом бланков для определения предела решимости и специфичности).
3.1.2.1. Восстановление
При отсутствии CRM восстановление определяется экспериментально, используя обогащенный образец матрицы согласно, например, следующему плану:
отбираются 18 аликвотных частей из одного образца материала и каждая группа из 6 аликвотных частей обогащается в 1, 1,5 и 2 раза требуемого минимального предела функционирования или в 0,5, 1 и 1,5 раза разрешенного предела;
исследуются пробы и определяется концентрация, присутствующая в каждой пробе;
при помощи указанного ниже уравнения подсчитывается восстановление для каждой пробы;
определяется среднее восстановление и CV для шести результатов от каждого уровня,
из чего следует, что % восстановления = 100Ч измеренное содержание/уровень обогащения.
Этот классический метод определения восстановления является разновидностью метода стандартных дополнений, описанного в пункте 3.5, если:
проба рассматривается как бланк (х1) вместо исследуемой пробы;
считается, что производительность и восстановление идентичны для обеих исследуемых долей (х2);
исследуемые пробы имеют ту же массу и экстракты исследуемых частей имеют те же объемы;
количество эталона, добавленного ко второй исследуемой части (обогащенной), обозначается xADD (xADD = ρA • VA), где:
x1 является измеренным значением бланка образца, а x2 – измеренным значением второй исследуемой (обогащенной) части,
откуда следует, что % восстановления = 100 (x2 – x1)/xADD.
Если любое из вышеуказанных условий не выполняется или предполагается, что не выполняется, необходимо выполнить полную процедуру для определения восстановления методом стандартных дополнений, описанным в пункте 3.5.
Примечание: Производительность: доля массы aналита, содержащаяся в последнем экстракте.
Восстановление (здесь): доля массы aналита, добавляемая к пробе, которая присутствует в последнем экстракте. В продолжение настоящего документа считается, что производительность и восстановление идентичны и используется только понятие «восстановление».
3.1.2.2. Повторяемость
Готовится набор проб идентичных матриц, обогащенных aналитом, таким образом, чтобы получить концентрации, равные 1, 1,5 и 2 требуемым минимальным пределом функционирования или 0,5, 1 и 1,5 разрешенным пределам.
На каждом уровне должны проводиться не менее шести повторных исследований.
Пробы исследуются.
Подсчитывается концентрация, обнаруженная в каждой пробе.
Определяются средняя концентрация, стандартное отклонение и коэффициент вариации (%) обогащенных проб.
Эти действия повторяются не менее двух раз.
Определяются общее число средних концентраций и CV для обогащенных проб.
3.1.2.3. Внутрилабораторная воспроизводимость
Из указанного материала для исследования готовится набор проб (идентичные или различные матрицы), обогащенных аналитом или аналитами таким образом, чтобы получить эквивалент в 1, 1,5 и 2 раза концентрации требуемого минимального предела функционирования или в 0,5, 1 и 1,5 раза разрешенного предела.
На каждом уровне следует проводить не менее шести исследований.
Эти действия повторяются не менее двух раз различными операторами, в различных условиях, например, различные партии реагентов, растворителей и т.д., при различных окружающих температурах, различными инструментами и т.д., по возможности.
Пробы исследуются.
Подсчитывается концентрация, обнаруженная в каждой пробе.
Определяются средняя концентрация, стандартное отклонение и коэффициент вариации (%) обогащенной пробы.
3.1.2.4. Воспроизводимость
В случае, если необходимо проверить воспроизводимость, лаборатории должны участвовать в исследованиях согласно стандарту ISO 5725-2.
3.1.2.5. Предел решимости (CCα)
Предел решимости устанавливается в соответствии с требованиями по идентификации или по идентификации и квантификации, установленными Критериями функционирования и другими требованиями, применяемыми к методам испытаний (часть 2).
Для веществ с неустановленным разрешенным пределом предел решимости может быть установлен:
либо методом градуировочной кривой в соответствии со Стандартом ISO 11843 (именуемой здесь критическим значением переменной среднего состояния). При этом используется несколько образцов проб, обогащенных до требуемого минимального уровня функционирования и выше этого уровня, с равным добавлением. Пробы исследуются. После идентификации сигнал представляется как функция добавленной концентрации. Предел решимости равен концентрации соответствующей добавки y плюс 2,33 раза стандартных отклонений внутрилабораторной воспроизводимости. Это применяется исключительно к количественным определениям (α = 1%);
либо путем исследования не менее 20 образцов материалов для каждой матрицы, чтобы определить соотношение сигнал-шум в окне, в котором ожидается аналит. В качестве предела решимости можно использовать величину, равную тройному соотношению сигнал-шум. Этот метод применяется к количественным и качественным определениям.
Для веществ с установленным разрешенным пределом, предел решимости может быть установлен:
либо методом градуировочной кривой (именуемой здесь критическим значением состояния нетто) в соответствии со стандартом ISO 11843. В данном случае используется несколько пробных образцов, обогащенных до уровня, близкого к разрешенному пределу с равной добавкой. Пробы исследуются. После идентификации сигнал представляется как функция добавленной концентрации. Предел решимости равен концентрации, соответствующей разрешенному пределу плюс 1,64 стандартных отклонений внутрилабораторной воспроизводимости (α = 5%);
либо путем исследования не менее 20 образцов материала матрицы, обогащенного аналитом или аналитами к разрешимому пределу. Предел решимости равен концентрации к разрешенному пределу плюс 1,64 соответствующих стандартных отклонений (α = 5%).
3.1.2.6. Способность обнаружения (CCβ)
Предел обнаружения определяется в соответствии с требованиями по определению, идентификации или идентификации и квантификации, указанными в части 2.
Для веществ с неустановленным разрешенным пределом предел решимости может быть определен:
методом градуировочной кривой (именуемой здесь минимальным обнаруживаемым переменной определенного состояния) согласно стандарту ISO 11843. При этом используется репрезентативный образец материала, обогащенный до требуемого минимального уровня функционирования и выше этого уровня, с равной добавкой. Пробы исследуются. После идентификации сигнал представляется как функция добавленной концентрации. Лимит обнаружения равен концентрации, соответствующей пределу решимости плюс 1,64 стандартных отклонений внутрилабораторной воспроизводимости для измеренного среднего содержания в пределе решимости (β = 5%);
путем анализа не менее 20 бланк-материалов для каждой матрицы, обогащенных аналитом или аналитами до предела решимости. Анализируются пробы и идентифицируются аналиты. Предел обнаружения равен значению предела решимости плюс 1,64 стандартного отклонения внутрилабораторной воспроизводимости для измеренного содержания (β = 5%);
при отсутствии количественных результатов способность обнаружения может определяться путем изучения обогащенного бланк-материала в пределах решимости и выше этих пределов. При этом способность обнаружения метода равна уровню концентрации, при которой остается не более 5% ложных соответствующих результатов. Следовательно, должно проводится не менее 20 исследований для не менее одного уровня концентрации в целях получения надежной основы для этого определения.
В случае веществ, для которых установлен авторизированный предел, способность обнаружения может быть определена:
либо методом градуировочной кривой (именуемой здесь минимальным обнаруживаемым значением переменной состояния нетто) в соответствии с стандартом ISO 11843. При этом используется репрезентативная проба-бланк, обогащенная до и более требуемого минимального функционального уровня, с равным добавлением. Исследуются пробы и идентифицируется аналит (аналиты). Подсчитывается стандартное отклонение среднего состава, измеряемого на уровне решимости. Способность обнаружения равняется соответствующей концентрации значения предела решимости плюс 1,64 стандартного отклонения внутрилабораторной воспроизводимости (β = 5 %),
либо путем исследования не менее 20 бланк-материалов для каждой матрицы, обогащенных аналитом или аналитами до предела решимости. Способность обнаружения равна значению предела решимости плюс 1,64 раза соответствующего стандартного отклонения (β = 5%).
3.1.2.7. Прочность (существенные изменения)
Метод испытания должен апробироваться в разных экспериментальных условиях, охватывающих разные виды, разные матрицы или разные условия для пробы. Внесенные изменения должны быть значимыми. Их значимость можно оценить с использованием, например, подхода Youden. Каждая характеристика функционирования должна быть определена для всех существенных изменений, для которых был доказан существенный эффект при проведении апробирования.
3.1.3. Валидация на основании других методов
При использовании других методов валидации, в протоколе валидации уточняются образец и основная стратегия с первоначальными требованиями, гипотезы и соответствующие формулы или, по меньшей мере, делается ссылка на них. В дальнейшем приводится один из методов замены. При использовании метода внутренней валидации, например, характеристики функционирования определяются таким образом, чтобы один и тот же метод валидации позволил признание существенных колебаний. Для этого необходимо установить экспериментальный план по валидации.
3.1.3.1. План испытания
В зависимости от числа видов и различных изученных факторов устанавливается план испытания. Следовательно, на первом этапе валидации метода изучаются популяции проб, которые затем исследуются в лаборатории для того, чтобы выбрать главные виды и факторы, которые могут повлиять на результаты измерений. Предел концентраций выбирается в зависимости от цели, в соответствии с учтенным уровнем.
Пример:
несколько аналитов может быть изучено одновременно в процессе валидации метода испытания;
идентифицированы два варианта основного фактора (A и B). Главные факторы составляют основу комбинирования уровней факторов. Эти главные факторы могут охватить такие факторы, как вид или матрица. В данном примере главный фактор изменился в двух плоскостях, то есть были рассмотрены два разных вида (вид A и вид B). В общем возможно, чтобы главные факторы были изменены более чем на два уровня, что позволяет увеличить число проведенных анализов;
выбранные факторы изменяются на два уровня (обозначенные + или -).
как важные для метода валидации
|
Пол животного
Вид
Условия перевозки
Условия хранения Условия откорма
Свежесть пробы Разные потребители, имеющие разный опыт |
(фактор 1)
(фактор 2)
(фактор 3)
(фактор 4)
(фактор 5)
(фактор 6)
(фактор 7)
|
план для вышеописанного примера
|
Вид
|
Фактор 1
|
Фактор 2
|
Фактор 3
|
Фактор 4
|
Фактор 5
|
Фактор 6
|
Фактор 7
|
Номер
пробы
|
|
A
A
A
A
A
A
A
A
B
B
B
B
B
B
B
B
|
+
+
+
+
-
-
-
-
+
+
+
+
-
-
-
-
|
+
+
-
-
+
+
-
-
+
+
-
-
+
+
-
-
|
+
-
+
-
+
-
+
-
+
-
+
-
+
-
+
-
|
+
-
-
+
-
+
+
-
+
-
-
+
-
+
+
-
|
-
+
-
+
+
-
+
-
+
-
+
-
-
+
-
+
|
+
-
-
+
+
-
-
+
-
+
+
-
-
+
+
-
|
-
-
+
+
+
+
-
-
+
+
-
-
-
-
+
+
|
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
|
Поскольку каждая проба (каждая комбинация уровней факторов) должна обогащаться четырьмя различными концентрациями, приближенными к интересующему уровню, а бланк-проба должна быть проанализирована для каждого уровня, следовательно, необходимо выполнить 5 х 16 = 80 анализов для набора исследований по валидации.
Эти 80 результатов измерений позволяют осуществить подсчет.
Восстановление:
повторяемость на уровне концентрации (Sir);
межлабораторная воспроизводимость на уровень концентрации (SiR);
предел решимости (CCa);
способность обнаружения (CCβ);
кривая силы (долю ошибок β в отношении с концентрацией см. в пункте 3.1.2.3);
прочность (существенные вариации); прочность с незначительными вариациями может быть определена согласно пункту 3.1.1.3;
16 градуировочных кривых, связанных с пробой;
одна градуировочная кривая;
интервал предсказания суммарной градуировочной кривой;
отклонения, наведенные матрицей (Smat);
отклонения, наведенные проведением исследования (Srun);
влияние различных факторов на результаты измерений.
Эти функциональные характеристики позволяют осуществить полную оценку уровня функционирования метода, поскольку изучается не только влияние разных факторов, но и комбинации этих факторов. С помощью этого плана испытаний можно принять решение об исключении одного или другого фактора, выбранного из общей градуировочной кривой, так как существенно отделяется от стандартных отделений остальных факторов.
3.1.3.2. Кривая силы
Кривая силы предоставляет информацию о способности обнаружения метода ряда выбранных концентраций. Она ссылается на риск искаженности β при использовании изучаемого метода. Кривая силы позволяет рассчитать способность обнаружения разных категорий (выявление, подтверждение) или типов (качественного или количественного) методов для допустимой искаженности β (например 5%).
Кривая силы
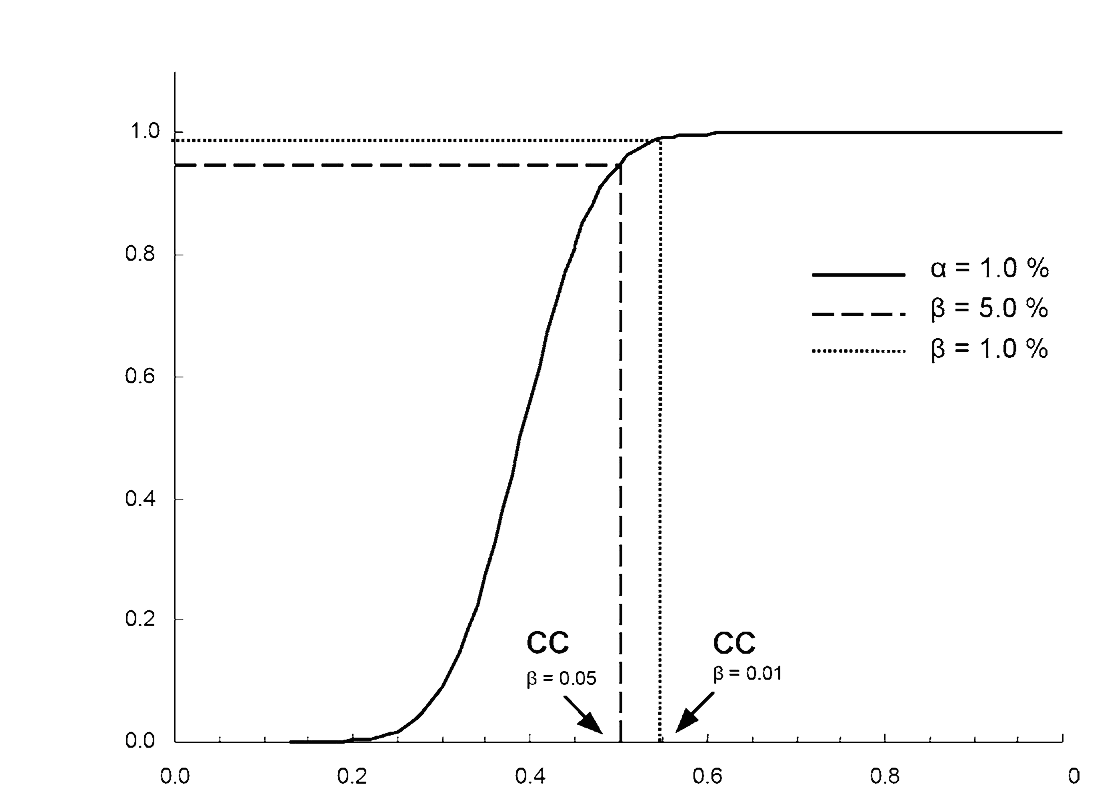
Рисунок 1 является примером графического представления способности обнаружения (ССβ) одного метода исследования. Этот метод имеет 5% ложной решимости. При концентрации 0,50 µг/кг риск ложности снижается до 1%.
3.1.3.3. Воспроизводимость
Определение воспроизводимости одного метода путем внутрилабораторного исследования (внутренней валидации) требует участия в тестах на опытность в соответствии со справочниками ISO 43-1 и 43-2. Лаборатории могут выбрать собственные методы при условии, что методы используются в повседневных условиях.
Стандартное отклонение результатов лаборатории может использоваться для оценки воспроизводимости метода.
3.2. Графическое представление различных пределов исследования
Вещества с неустановленным авторизированным пределом
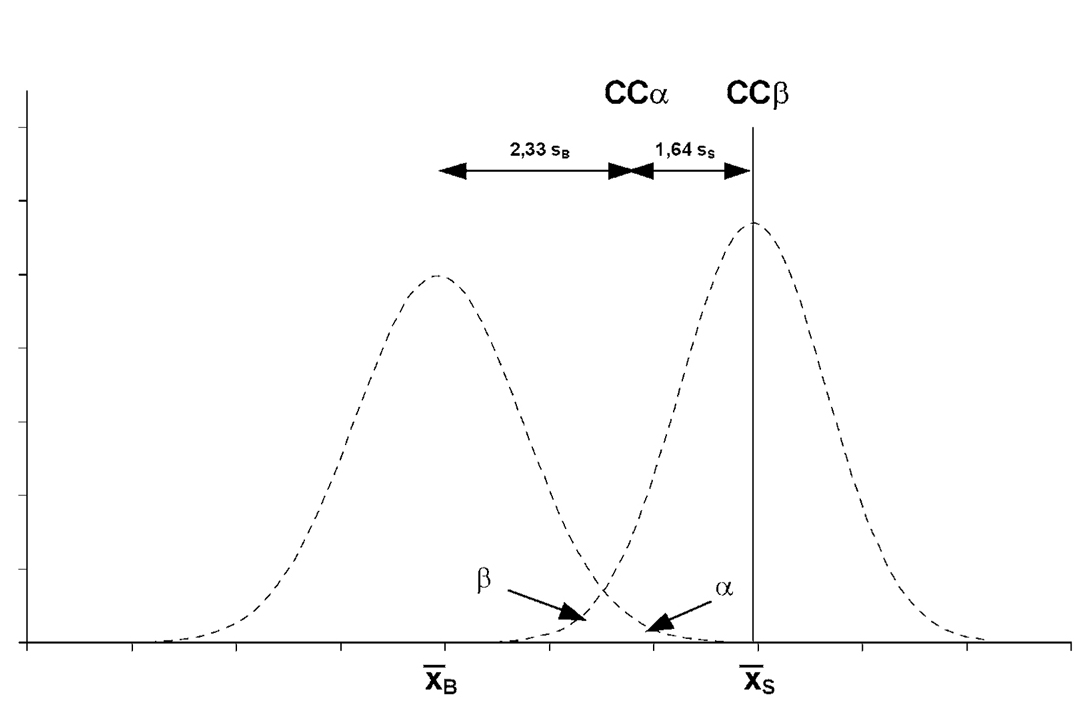
Sg - стандартное отклонение оценки результатов пробы-бланк (определенное в условиях внутрилабораторной воспроизводимости);
Ss - стандартное отклонение оценки результатов зараженной пробы (определенная в условиях внутрилабораторной воспроизводимости);
α - норма ложнонесоответствующих результатов;
β - норма ложносоответствующих результатов;
CCα - ответ с искажением α и искажением β в 50%;
CCβ - ответ с минимальным искажением α и данным искажением β.
Вещества с авторизированными пределами
Авторизированный предел (PL)
Частота CC α CCβ
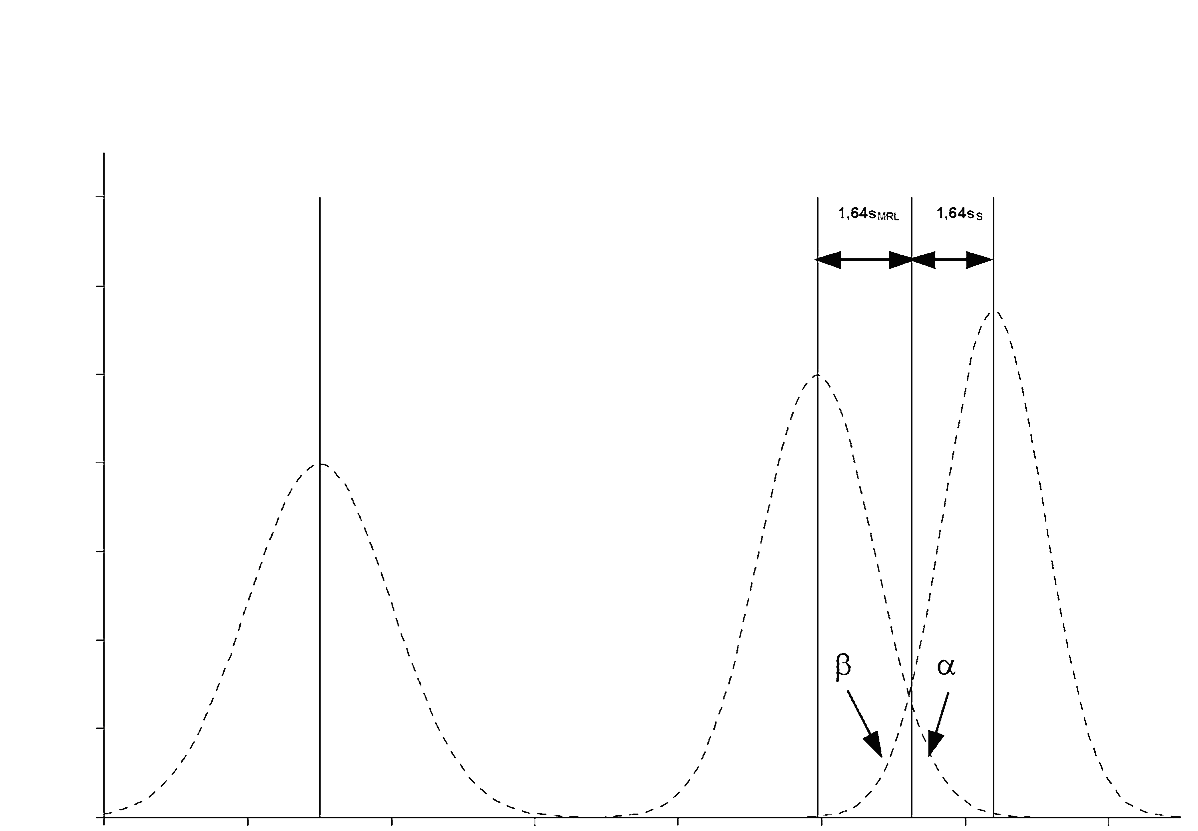
Концентрация
X g - средняя концентрация бланк-пробы;
X PL - средняя концентрация пробы, содержащей авторизированный предел аналита;
X c - средняя концентрация зараженной пробы;
SPL - стандартное отклонение пробы, содержащей авторизированный предел аналита (определенное в условиях внутрилабораторной воспроизводимости);
SS - стандартное отклонение зараженной пробы (определенное в условиях межлабораторной воспроизводимости);
α - норма ложнонесоответствующих результатов;
β - норма ложносоответствующих результатов;
CC α - ответ с искажением α и искажением β в 50%;
CC β - ответ с минимальным искажением α и данным искажением β.
3.3. Пример решения для определения верности метода при минимальных отклонениях по Youden.
Среднее сравнение (A)
|
AA = Σ(Ai)/4
AB = Σ(Bi)/4
AC = Σ(Ci)/4 |
Сравниваются средние прописных (от АА до AG) со средними строчных соответствующих (от Аа до Ag). В случае, если один фактор имеет какой-либо эффект, разница будет существенно больше чем разницы других факторов |
|
AD = Σ(Di)/4
AE = Σ(Ei)/4
|
Массивный метод не должен быть под воздействием колебаний, встречающихся с квазиуверенностью между лабораториями |
|
AF = Σ(Fi)/4
AG = Σ(Gi)/4
Aa = Σ(ai)/4
Ab = Σ(bi)/4
Ac = Σ(ci)/4
Ad = Σ(di)/4
Ae = Σ(ei)/4
Af = Σ(fi)/4
Ag = Σ(gi)/4 |
В отсутствие особой разницы, самая реалистическая оценка случайной ошибки отображается 7 разницами |
|
Разница (Di)
|
Квадраты разница, Di2
|
|
Da = A – a = Σ(Ai) – Σ(ai) Db = B – b = Σ(Bi) – Σ(bi)
Dc = C – c = Σ(Ci) – Σ(ci)
Dd = D – d = Σ(Di) – Σ(di) De = E – e = Σ(Ei) – Σ(ei)
Df = F – f = Σ(Fi) – Σ(fi)
Dg = G – g = Σ(Gi) – Σ(gi) |
Da2 = значение a
Db2 = значение b Dc2 = значение c Dd2 = значение d De2 = значение e Df2 = значение f Dg2 = значение g |
Стандартное отклонение разниц Di (SDi):
3.4. Примеры подсчета для процедуры внутреннего признания
Примеры и подсчеты для протокола внутреннего признания, описанного в пункте 3.1.3. Признание, основанное на других методах.
3.5. Примеры подсчета для метода эталонных добавок
Проба для исследования, содержащая Т-аналит, делится на две массовые фракции – 1 и 2 (m1 и m2). Фракция 2 обогащается раствором в объеме VA с концентрацией аналита ρA. После экстрагирования и очищения метода получаются два экстракта из этих фракций с объемами V1 и V2. Считается, что возмещение аналита это rc. Полученные два экстракта исследуются методом с чувствительностью b и получаются аналитические результаты x1 и x2 соответственно.
Предполагая, что rc и b являются идентичными для аналита в первоначальной и обогащенной пробах, содержимое Т вычисляется следующим образом:
Также возможно вычислить чувствительность b следующим образом: