ID intern unic: 376983
Версия на русском

Republica Moldova
din 11.07.2018
pentru aprobarea Regulamentului privind condițiile
de introducere pe piață a dispozitivelor medicale
pentru diagnostic in vitro
de introducere pe piață a dispozitivelor medicale
pentru diagnostic in vitro
În temeiul art. 10 alin. (1) din Legea nr.102 din 9 iunie 2017 cu dispozitivele cu privire la dispozitivele medicale (Monitorul Oficial al Republicii Moldova, 2017, nr. 244-251, art. 389), cu modificările ulterioare, Guvernul HOTĂRĂȘTE
1. Se aprobă Regulamentul privind condițiile de introducere pe piață a dispozitivelor medicale pentru diagnostic in vitro (se anexează)
2. Se admite introducerea pe piață și punerea în funcțiune a dispozitivelor medicale pentru diagnostic in vitro care poartă marcajul de conformitate SM, aplicat conform prevederilor din Legea nr. 235 din 1 decembrie 2011 privind activitățile de acreditare și de evaluare a conformității, și sînt însoțite de declarația de conformitate.
Producătorul sau reprezentantul autorizat al acestuia, persoană juridică cu sediul în Republica Moldova, aplică marcajul de conformitate SM în situația în care evaluarea conformității dispozitivelor medicale pentru diagnostic in vitro se realizează de către organismele de evaluare a conformității notificate prin utilizarea procedurilor prevăzute de Regulamentul privind condițiile de introducere pe piață a dispozitivelor medicale pentru diagnostic in vitro.
Se interzice, în condițiile prevăzute de Regulamentul menționat, aplicarea pe același dispozitiv a marcajului de conformitate SM și a marcajului CE.
3. Ministerul Sănătății, Muncii și Protecției Sociale recunoaște organismele acreditate conform prevederilor din Legea nr. 235 din 1 decembrie 2011 privind activitățile de acreditare și de evaluare a conformității, care realizează evaluarea conformității dispozitivelor medicale pentru diagnostic in vitro destinate pieței naționale în concordanță cu procedurile prevăzute în Regulamentul privind condițiile de introducere pe piață a dispozitivelor medicale pentru diagnostic in vitro.
4. Cerințele cu privire la organismele de evaluare a conformității notificate se aplică și organismelor de evaluare a conformității recunoscute. La desfășurarea procedurilor de evaluare a conformității, organismele de evaluare a conformității recunoscute care realizează evaluarea conformității dispozitivelor medicale pentru diagnostic in vitro vor întocmi certificate de examinare de tip.
5. Lista cuprinzînd organismele de evaluare a conformității recunoscute, sarcinile specifice pentru care acestea au fost recunoscute și numerele lor de identificare se publică în Monitorul Oficial al Republicii Moldova.
6. Obligațiile și răspunderea producătorului, reprezentantului său autorizat, ale importatorului sau distribuitorului, persoane juridice cu sediul în Republica Moldova, privind dispozitivele medicale pentru diagnostic in vitro puse în funcțiune care dețin marcajul de conformitate SM sînt similare cu cele prevăzute de prezenta hotărîre pentru dispozitivele medicale pentru diagnostic in vitro care dețin marcajul CE.
7. Toate activitățile ce țin de controlul de stat sau activitățile de supraveghere a pieței care înglobează în sine activitatea de control de stat, în special aplicarea măsurilor de interzicere sau restrîngere a plasării dispozitivelor medicale pentru diagnostic in vitro și activitățile ce țin de controlul de stat al dispozitivelor medicale pentru diagnostic in vitro de pe piață prevăzute de prezentul Regulament vor fi efectuate de către Agenția Națională pentru Sănătate Publică, în comun cu Agenția Medicamentului și Dispozitivelor Medicale, în conformitate cu prevederile Legii nr. 131 din 8 iunie 2012 privind controlul de stat asupra activității de întreprinzător și ale Legii nr.102 din 9 iunie 2017 cu privire la dispozitivele medicale.
8. Controlul asupra executării prezentei hotărîri se pune în sarcina Ministerului Sănătății, Muncii și Protecției Sociale.
9. Se abrogă Hotărîrea Guvernului nr. 435 din 10 iunie 2014 „Pentru aprobarea Regulamentului privind condițiile de plasare pe piață a dispozitivelor medicale pentru diagnostic in vitro” (Monitorul Oficial al Republicii Moldova, 2014, nr. 160-166, art. 482).
PRIM-MINISTRU Pavel FILIP
Contrasemnează:
Ministrul sănătății,
muncii și protecției sociale Svetlana Cebotari
Ministrul economiei și infrastructurii Chiril Gaburici
Ministrul afacerilor externe
și integrării europene Tudor Ulianovschi
Ministrul justiției Victoria Iftodi
Nr. 703. Chişinău, 11 iulie 2018.
2. În sensul prezentului Regulament se utilizează terminologia definită în Legea nr. 102 din 9 iunie 2017 cu privire la dispozitivele medicale, Legea nr. 235 din 1 decembrie 2011 privind activitățile de acreditare și de evaluare a conformității, Legea nr. 1409-XIII din 17 decembrie 1997 cu privire la medicamente, precum și următoarele noțiuni:
dispozitiv pentru autotestare – orice dispozitiv conceput de către producător astfel încât să fie folosit de nespecialiști în condiții casnice;
producător – persoană fizică sau juridică care are responsabilitatea de a proiecta, a produce, a ambala și a eticheta un dispozitiv medical pentru introducerea lui pe piață sub propria sa denumire, indiferent dacă această operațiune este efectuată de această persoană personal sau de o parte terță în numele ei (responsabilă de introducerea pe piață). Noțiunea se aplică și persoanelor fizice sau juridice care asamblează, ambalează, prelucrează, recondiționează și/sau etichetează produse și/sau atribuie acestora destinația de dispozitiv medical, cu intenția de introducere a acestuia pe piață sub propria sa denumire. Noțiunea nu se aplică persoanelor care, nefiind producători în înțelesul acestei definiții, asamblează sau adaptează dispozitive medicale deja existente pe piață pentru un anumit pacient;
dispozitiv de evaluare a performanței – dispozitiv destinat de către producător să fie supus unuia sau mai multor studii de evaluare a performanței în laboratoare pentru analize medicale sau într-un alt spațiu adecvat aflat în afara propriei sale incinte.
3. În sensul prezentului Regulament, materialele de control și etalon includ orice substanță, material sau articol destinat de producător fie să stabilească relații de măsurare, fie să calibreze sau să verifice caracteristicile de performanță ale unui dispozitiv în legătură cu scopul acestuia.
4. În sensul prezentului Regulament, îndepărtarea, recoltarea și utilizarea țesuturilor, a celulelor și a substanțelor de origine umană sînt reglementate prin Legea nr. 42-XVI din 6 martie 2008 privind transplantul de organe, țesuturi și celule umane.
5. Prevederile prezentului Regulament nu se aplică dispozitivelor fabricate și utilizate numai în cadrul aceleiași unități medicale și, respectiv, la sediul producătorului sau utilizate în clădiri din imediata vecinătate, fără să fi fost transferate unei alte persoane juridice. Aceasta nu va afecta dreptul Agenției Medicamentului și Dispozitivelor Medicale (în continuare – Agenție) de a supune astfel de activități unor cerințe specifice de protecție.
6. Prevederile prezentului Regulament nu aduc atingere actelor normative în vigoare privind furnizarea de dispozitive pe bază de prescripție medicală.
8. Dispozitivele trebuie să îndeplinească cerințele esențiale prevăzute în anexa nr. 1 la prezentul Regulament și care le sînt aplicabile, conform scopului pentru care au fost proiectate respectivele dispozitive.
10. Pentru dispozitivele destinate evaluării performanței pentru laboratoarele și instituțiile indicate în declarația prevăzută în anexa nr. 8 la prezentul Regulament nu sînt restricții de introducere pe teritoriul Republicii Moldova dacă satisfac condițiile prevăzute în prezentul Regulament.
11. Prevederile prezentului Regulament nu se aplică dispozitivelor destinate expunerii în cadrul tîrgurilor, expozițiilor, demonstrațiilor, întrunirilor științifice și tehnice organizate pe teritoriul Republicii Moldova, cu condiția ca acestea să nu fie utilizate pentru efectuarea analizelor pe probe prelevate de la participanți și să existe o indicație vizibilă că aceste dispozitive nu pot fi comercializate sau puse în funcțiune înainte de a deveni conforme cu prezentul Regulament.
12. Pentru asigurarea unei utilizări sigure și corecte a dispozitivului, informațiile prevăzute în anexa nr. 1 partea В, secțiunea a 9-a se furnizează în limba de stat a Republicii Moldova.
13. La aplicarea prevederilor pct. 12 din prezentul Regulament se ține cont de principiul proporționalității, în mod special:
1) de faptul dacă informațiile trebuie să fie furnizate prin simboluri armonizate, coduri sau alte măsuri notificate;
2) de tipul de utilizator anticipat pentru respectivul dispozitiv.
14. Înregistrările și corespondența privind aplicarea procedurilor de evaluare a conformității se prezintă în limba de stat sau într-o limbă acceptată de organismul notificat de evaluare a conformității.
15. În cazul în care dispozitivele intră sub incidența altor reglementări care, de asemenea, prevăd aplicarea marcajului de conformitate, acesta trebuie să indice conformitatea dispozitivelor cu prevederile tuturor reglementărilor aplicabile.
16. Dacă una sau mai multe reglementări menționate la pct. 15 din prezentul Regulament permit producătorului, pentru o perioadă tranzitorie, să aleagă reglementările pe care să le aplice, marcajul CE semnifică faptul că dispozitivele satisfac numai prevederile acelor reglementări tehnice care sînt aplicate de producător.
17. În acest caz, elementele de identificare particulare ale reglementărilor tehnice aplicate trebuie să fie indicate în documentele, notele sau instrucțiunile care însoțesc dispozitivul.
18. Se consideră că cerințele esențiale prevăzute în anexa nr. 1 la prezentul Regulament sînt îndeplinite dacă dispozitivele medicale sînt conforme cu specificațiile tehnice din standardele europene armonizate, adoptate în calitate de standarde moldovenești din domeniul dispozitivelor medicale (în continuare – standarde).
19. Lista standardelor din domeniul dispozitivelor medicale pentru diagnostic in vitro se aprobă prin ordin de către Agenție și se publică în Monitorul Oficial al Republicii Moldova. Această listă se actualizează ori de cîte ori este cazul.
20. În situația în care Agenția constată că standardele nu respectă în întregime cerințele esențiale prevăzute în anexa nr. 1 la prezentul Regulament, aceasta aplică prevederile prezentului Regulament.
21. Se consideră conforme cu cerințele esențiale stipulate în anexa nr. 1 la prezentul Regulament dispozitivele medicale pentru diagnostic in vitro proiectate și fabricate în conformitate cu specificațiile tehnice generale elaborate pentru dispozitivele incluse în listele A și B din anexa nr. 2 la prezentul Regulament. Specificațiile tehnice generale sînt aprobate prin ordin al ministrului sănătății, muncii și protecției sociale și stabilesc criteriile de evaluare și de reevaluare a performanței, criteriile de eliberare a loturilor, metodele și materialele de referință.
22. Producătorii trebuie să se conformeze specificațiilor tehnice generale sau, după caz, să asigure condiții echivalente specificațiilor tehnice generale în vigoare. Standardele cu referire la dispozitivele medicale pentru diagnostic in vitro includ și specificațiile tehnice generale.
24. Agenția informează părțile interesate de măsurile întreprinse în conformitate cu prevederile Legii nr. 102 din 9 iunie 2017 cu privire la dispozitivele medicale, invocînd motive întemeiate, în mod special dacă neconformitatea cu prezentul Regulament se datorează:
1) neîndeplinirii cerințelor esențiale prevăzute în anexa nr. 1 la prezentul Regulament;
2) aplicării incorecte a standardelor prevăzute la pct. 18 din prezentul Regulament, în măsura în care se pretinde că standardele au fost aplicate;
3) unor deficiențe ale standardelor.
25. Dacă un dispozitiv neconform poartă marcajul CE, Agenția ia măsurile corespunzătoare în conformitate cu prevederile Legii nr. 102 din 9 iunie 2017 cu privire la dispozitivele medicale.
27. În cazul dispozitivelor medicale pentru autotestare, înainte de a emite declarația de conformitate menționată la pct. 26 din prezentul Regulament, producătorul trebuie să îndeplinească cerințele suplimentare stabilite în pct. 6 din anexa nr. 3 la prezentul Regulament. În locul acestei proceduri, producătorul poate aplica procedura prevăzută la pct. 29 sau 30 din prezentul Regulament.
28. În cazul tuturor dispozitivelor cuprinse în lista A din anexa nr. 2 la prezentul Regulament, cu excepția celor destinate evaluării performanței, producătorul, în scopul aplicării marcajului CE, recurge la una dintre următoarele proceduri referitoare la:
1) declarația de conformitate CE stabilită în anexa nr. 4 la prezentul Regulament;
2) examinarea CE de tip stabilită în anexa nr. 5, asociată cu procedura referitoare la declarația de conformitate, conform anexei nr. 7 la prezentul Regulament privind asigurarea calității producției.
29. În cazul tuturor dispozitivelor cuprinse în lista B din anexa nr. 2 la prezentul Regulament, cu excepția celor destinate evaluării performanței, producătorul, în scopul aplicării marcajului de conformitate, apelează la una dintre următoarele proceduri referitoare la:
1) declarația de conformitate CE stabilită în anexa nr. 4 la prezentul Regulament;
2) examinarea CE de tip stabilită în anexa nr. 5 la prezentul Regulament, asociată cu:
a) procedura privind verificarea CE stabilită în anexa nr. 6 la prezentul Regulament;
b) procedura referitoare la declarația de conformitate CE menționată în anexa nr. 7 la prezentul Regulament.
30. În cazul dispozitivelor de evaluare a performanței, producătorul aplică procedura stabilită în anexa nr. 8 la prezentul Regulament și emite declarația prevăzută în această anexă înainte ca dispozitivele respective să devină disponibile.
Dispozițiile prezentului punct nu afectează reglementările naționale referitoare la aspectele etice ale studiilor de evaluare a performanței dispozitivelor pentru a căror fabricare sînt folosite țesuturi sau substanțe de origine umană.
31. În timpul procedurii de evaluare a conformității unui dispozitiv, producătorul și, dacă este implicat, organismul de certificare notificat trebuie să țină cont de rezultatele obținute în urma oricăror operațiuni de evaluare și verificare efectuate, după caz, în concordanță cu prezentul Regulament, într-o fază intermediară de fabricație.
32. Producătorul poate da instrucțiuni reprezentantului său autorizat în sensul începerii procedurilor prevăzute în anexele nr. 3, nr. 5, nr. 6 și nr. 8 din prezentul Regulament.
33. Producătorul trebuie să păstreze timp de 5 ani de la data fabricării ultimului produs declarația de conformitate, documentația tehnică menționată în anexele nr. 3-8 la prezentul Regulament, precum și hotărîrile, rapoartele și certificatele emise de organismele notificate și, în caz de necesitate, să le pună la dispoziția Agenției pentru inspectare.
Dacă producătorul nu este stabilit în Republica Moldova, obligația de a pune la dispoziție, la cerere, documentația menționată la primul alineat al prezentului punct îi revine reprezentantului său autorizat.
34. Atunci cînd procedura de evaluare a conformității implică intervenția unui organism de certificare notificat, producătorul ori reprezentantul său autorizat se adresează unui organism la alegere, corespunzător sarcinilor în legătură cu care acesta a fost notificat.
35. Organismul notificat cere, în cazurile întemeiate, orice informații sau date necesare pentru stabilirea și menținerea atestării conformității în funcție de procedura aleasă.
36. Deciziile adoptate de organismul notificat în conformitate cu anexele nr. 3-nr.5 la prezentul Regulament au valabilitatea stabilită în Legea nr. 235 din 1 decembrie 2011 privind activitățile de acreditare și de evaluare a conformității.
37. Înregistrările și corespondența referitoare la procedurile prevăzute la pct. 26-29 din prezentul Regulament se redactează în limba de stat.
38. Agenția poate permite, în baza unor informații justificate, înregistrarea în Registrul de stat al dispozitivelor medicale, în scopul introducerii pe piață și al punerii în funcțiune în Republica Moldova, a dispozitivelor medicale pentru care nu s-au efectuat procedurile de evaluare a conformității și a căror utilizare este în interesul protecției sănătății.
39. Prevederile prezentului capitol se aplică în mod corespunzător oricărei persoane fizice sau juridice care produce dispozitivele ce fac obiectul prezentului Regulament și care, fără să le introducă pe piață, le pune în funcțiune și le utilizează în cadrul activității sale profesionale.
1) adresa sediului juridic/domiciliul;
2) informațiile despre reactivi, produși de reacție și materiale de calibrare și control, în ceea ce privește caracteristicile tehnologice generale și/sau substanțele de analizat și orice modificare semnificativă a acestora, inclusiv suspendarea introducerii pe piață;
3) în cazul dispozitivelor care fac obiectul anexei nr. 2 la prezentul Regulament și al dispozitivelor de autotestare, parametrii analitici și, după caz, parametrii de diagnostic conform anexei nr. 1 partea A pct. 3, rezultatele evaluării performanței conform anexei nr. 8 la prezentul Regulament, datele despre certificate și orice modificări semnificative ale acestora, inclusiv sistarea introducerii pe piață a dispozitivelor.
1) orice disfuncție sau orice alterare/depreciere a caracteristicilor și/sau a performanțelor unui dispozitiv medical pentru diagnostic in vitro, precum și orice etichetare, prospect și instrucțiune inadecvate, susceptibile să producă sau că au produs, direct ori indirect, decesul sau afectarea severă a stării de sănătate a unui pacient ori utilizator;
2) orice argument de ordin tehnic sau medical în legătură cu caracteristicile ori performanțele unui dispozitiv la care se referă subpct. 1) din prezentul punct și care ar conduce la o retragere sistematică de pe piață de către producător a dispozitivelor de același tip.
42. Obligația de a anunța Agenția despre incidentele în utilizarea dispozitivelor specificate în prezentul Regulament îi revine producătorilor, reprezentanților autorizați, utilizatorilor, persoanelor juridice ce comercializează dispozitive medicale și importatorilor în conformitate cu prevederile Legii nr. 102 din 9 iunie 2017 cu privire la dispozitivele medicale.
43. După efectuarea unei evaluări, dacă este posibil, împreună cu producătorul și fără a prejudicia aplicarea prevederilor pct. 23 din prezentul Regulament, Agenția informează părțile interesate cu privire la incidentele specificate în prezentul Regulament, pentru care s-au luat sau trebuie să se ia măsuri corespunzătoare, în conformitate cu prevederile Legii nr. 102 din 9 iunie 2017 cu privire la dispozitivele medicale, inclusiv retragerea.
44. Agenția informează, la cerere, autoritățile competente din alte state, cu care are încheiate acorduri cu privire la aspectele menționate la pct. 41-43 din prezentul Regulament și stabilește procedurile de aplicare a prevederilor din prezenta secțiune.
45. Datele înregistrate sînt stocate în baza de date, astfel încît să permită Agenției îndeplinirea atribuțiilor sale conform prezentului Regulament.
Normele de procedură pentru aplicarea prevederilor prezentului punct se aprobă prin ordin al ministrului sănătății, muncii și protecției sociale.
46. Baza de date va cuprinde:
1) datele referitoare la înregistrarea producătorilor și a dispozitivelor, potrivit secțiunii 1 a prezentului capitol;
2) datele referitoare la certificatele emise, modificate, suplimentate, suspendate, retrase sau respinse conform procedurilor prevăzute în anexele nr. 3-nr. 7 la prezentul Regulament;
3) datele obținute potrivit procedurii de vigilență prevăzute în prezenta secțiune la prezentul capitol.
Datele menționate se furnizează în format standard.
47. Procedurile pentru punerea în aplicare a prezentului capitol se adoptă în conformitate cu procedura de reglementare prevăzută de prezentul Regulament.
49. Agenția informează părțile interesate despre măsurile aplicate conform prevederilor din prezentul Regulament.
50. Măsurile necesare pentru modificarea elementelor neesențiale ale prezentului Regulament se adoptă în conformitate cu procedura de reglementare prevăzută de prezentul Regulament.
52. Ministerul Sănătății, Muncii și Protecției Sociale poate retrage recunoașterea unui organism în cazul în care constată că acest organism nu mai respectă criteriile menționate în anexa nr. 9 la prezentul Regulament.
53. Evaluarea conformității dispozitivelor medicale se efectuează de organisme de evaluare a conformității acreditate în condițiile Legii nr. 235 din 1 decembrie 2011 privind activitățile de acreditare și de evaluare a conformității și recunoscute de Ministerul Sănătății, Muncii și Protecției Sociale conform criteriilor stabilite de cadrul normativ în vigoare.
54. Organismul notificat și producătorul ori reprezentantul său autorizat stabilesc, de comun acord, termenele-limită pentru finalizarea activităților de evaluare și verificare prevăzute în anexele nr. 3-7 la prezentul Regulament.
55. Organismul notificat informează celelalte organisme și Ministerul Sănătății, Muncii și Protecției Sociale despre toate certificatele suspendate sau retrase și, la cerere, despre certificatele emise ori respinse. La cererea Ministerului Sănătății, Muncii și Protecției Sociale, pune la dispoziția lor toată informația suplimentară relevantă.
56. În cazul în care se constată că cerințele din prezentul Regulament nu au fost îndeplinite sau au încetat să mai fie îndeplinite de către producător ori dacă un certificat nu ar fi trebuit să fie emis, organismul notificat, ținînd cont de principiul proporționalității, suspendă sau retrage certificatul emis ori impune restricții asupra producătorului pînă în momentul în care conformitatea cu aceste cerințe este asigurată.
57. În cazul suspendării sau retragerii certificatului ori al impunerii de restricții asupra producătorului ori în cazurile în care este necesară o intervenție din partea Ministerului Sănătății, Muncii și Protecției Sociale, organismul notificat informează Ministerul Sănătății, Muncii și Protecției Sociale cu privire la acest fapt.
58. Organismul notificat furnizează, la cerere, toată informația și documentele relevante, pentru a da posibilitate Ministerului Sănătății, Muncii și Protecției Sociale să verifice îndeplinirea cerințelor prevăzute în anexa nr. 9 la prezentul Regulament.
60. Marcajul CE, conform anexei nr. 10 la prezentul Regulament, trebuie să fie aplicat într-o formă vizibilă, clară și care nu se poate șterge, atît pe dispozitiv sau pe ambalajul său steril, unde este posibil, cît și pe instrucțiunile de utilizare:
1) marcajul CE trebuie să fie aplicat și pe ambalajul în care se comercializează dispozitivul;
2) marcajul CE este însoțit de numărul de identificare al organismului notificat care poartă răspunderea pentru aplicarea procedurilor prevăzute în anexele nr. 3, nr. 4, nr. 6 și nr. 7 la prezentul Regulament.
61. Este interzisă aplicarea de simboluri sau de inscripții care pot induce în eroare terțe părți cu privire la înțelesul ori forma grafică a marcajului CE. Se admite aplicarea oricărui alt semn pe dispozitiv, pe ambalajul său ori pe instrucțiunile care însoțesc dispozitivul, cu condiția ca acesta să nu afecteze vizibilitatea și claritatea marcajului CE.
63. Dacă se menține situația de neconformitate prevăzută la pct. 62 din prezentul Regulament, Agenția adoptă toate măsurile necesare, în conformitate cu prevederile Legii nr. 102 din 9 iunie 2017 cu privire la dispozitivele medicale, pentru restrîngerea sau interzicerea introducerii pe piață a dispozitivului în cauză și/sau, după caz, pentru retragerea lui de pe piață.
64. Dispozițiile pct. 62 și 63 din prezentul Regulament se aplică și în cazul în care marcajul CE s-a aplicat inadecvat pe produse care nu fac obiectul prezentului Regulament.
66. Prevederile pct. 65 din prezentul Regulament nu se referă la obligațiile ce le revin Agenției și organismelor notificate cu privire la informarea reciprocă și difuzarea avertismentelor sau obligațiilor persoanelor implicate în furnizarea informațiilor sub incidența legislației penale în vigoare.
2. Soluțiile adoptate de către producător pentru proiectarea și fabricarea dispozitivelor trebuie să fie conforme cu principiile de siguranță, ținîndu-se cont de nivelul actual de dezvoltare a tehnologiei în domeniu. Pentru selectarea celor mai potrivite soluții, producătorul va aplica următoarele principii:
1) eliminarea sau reducerea la maximum a riscurilor prin proiectare și realizare sigură;
2) asigurarea măsurilor de protecție, acolo unde este cazul, în legătură cu riscurile care nu pot fi eliminate;
3) informarea utilizatorilor despre riscurile persistente, datorate insuficientelor măsuri de protecție adoptate.
3. Dispozitivele trebuie să fie proiectate și realizate în așa fel încît să fie adecvate scopurilor specificate de către producător, avînd în vedere nivelul actual de dezvoltare a tehnologiei în domeniu. Dispozitivele trebuie să realizeze următoarele performanțe: sensibilitate analitică, sensibilitate de diagnostic, specificitate analitică, specificitate de diagnostic, acuratețe, repetabilitate, reproductibilitate, inclusiv controlul interferențelor cunoscute și al limitelor de detecție stabilite de către producător. Trasabilitatea valorilor atribuite etaloanelor și/sau materialelor de control se asigură prin proceduri de măsurare de referință și/sau materiale de referință disponibile de calitate superioară.
4. Caracteristicile și performanțele specificate la pct. 1 și 3 din prezenta anexă nu trebuie să fie afectate în asemenea măsură încît să compromită sănătatea sau siguranța pacientului, a utilizatorului și, după caz, a altor persoane, pe întreaga durată de funcționare indicată de producător, atunci cînd dispozitivul este supus unor suprasolicitări ce pot interveni în timpul funcționării în condiții normale de exploatare. Dacă nu se specifică durata de funcționare, prevederea se aplică pentru durata de utilizare apreciată pentru un dispozitiv de acel tip, avînd în vedere scopul propus și utilizarea prevăzută a dispozitivului.
5. Dispozitivele trebuie să fie proiectate, fabricate și ambalate astfel încît caracteristicile și performanțele lor în timpul folosirii să nu fie afectate ca urmare a condițiilor de transport și de depozitare (temperatură, umiditate etc.), ținînd cont de instrucțiunile și informațiile furnizate de producător.
7. Dispozitivele trebuie să fie proiectate, fabricate și ambalate astfel încît să minimizeze riscul produs de scurgeri și contaminări cu reziduuri pentru persoanele implicate în transportul, depozitarea și utilizarea acestora, conform scopului propus.
9. Dacă un dispozitiv încorporează o substanță biologică, riscul de infecție trebuie să fie redus la minimum prin selectarea de donatori adecvați și de substanțe corespunzătoare, precum și prin utilizarea de proceduri adecvate validate de inactivare, conservare, testare și control.
10. Dispozitivele etichetate ca „STERILE” sau într-o stare microbiologică specială trebuie să fie proiectate, fabricate și ambalate pentru a se asigura păstrarea lor în starea microbiologică indicată pe etichetă atunci cînd sînt introduse pe piață, în condițiile de transport și depozitare specificate de către producător, dacă ambalajul nu este deschis sau deteriorat.
11. Dispozitivele etichetate ca „STERILE” sau într-o stare microbiologică specială trebuie să fie fabricate printr-o metodă corespunzătoare validată.
12. Sistemele de ambalare pentru dispozitive, altele decît cele menționate la pct. 10 din prezenta anexă, trebuie să păstreze produsul fără a-l deteriora, la nivelul de curățenie indicat de producător și, dacă dispozitivele trebuie să fie sterilizate înainte de utilizare, să reducă la minimum riscul de contaminare microbiană.
Este necesar să se ia măsuri pentru a se reduce la minimum riscul de contaminare microbiană în cursul selecției și al mînuirii de materii prime, al fabricării, al depozitării și al distribuției, dacă performanța dispozitivului este afectată de o astfel de contaminare.
13. Dispozitivele destinate sterilizării trebuie să fie fabricate în condiții (de exemplu, de mediu) corespunzătoare.
14. Sistemele de ambalare pentru dispozitivele nesterile trebuie să protejeze produsul de deteriorări, păstrîndu-se nivelul de curățenie prevăzut. La dispozitivele ce trebuie să fie sterilizate anterior folosirii trebuie redus riscul de contaminare microbiană; sistemul de ambalare trebuie să fie adecvat, conform metodei de sterilizare indicate de producător.
16. Dispozitivele trebuie să fie proiectate și fabricate astfel încît să reducă la minimum riscurile legate de utilizarea lor împreună cu materiale, substanțe și gaze cu care ar putea veni în contact în condiții normale de utilizare.
17. Dispozitivele trebuie să fie proiectate și fabricate astfel încît să elimine sau să reducă cît mai mult posibil:
1) riscul de lezare legat de caracteristicile fizice, inclusiv creșterile de volum/presiune/dimensiuni și caracteristicile ergonomice;
2) riscurile legate de influențe externe previzibile, cum ar fi: cîmpurile magnetice, efectele electrice externe, descărcările electrostatice, temperatura, umiditatea, presiunea și variațiile de presiune sau de accelerație ori pătrunderile accidentale de substanțe în dispozitiv.
Dispozitivele trebuie să fie proiectate și fabricate astfel încît să ofere un nivel adecvat de imunitate intrinsecă la perturbațiile electromagnetice, pentru a putea acționa conform scopului propus.
18. Dispozitivele trebuie să fie proiectate și fabricate în așa fel încît să reducă riscurile de incendiu sau explozie în timpul folosirii normale și în condiții de defectare simplă. O atenție deosebită trebuie să fie acordată dispozitivelor a căror utilizare proiectată presupune expunerea la sau folosirea lor în asociație cu substanțe inflamabile ori substanțe care întrețin arderea.
19. Dispozitivele trebuie să fie proiectate și fabricate astfel încît să faciliteze distrugerea în siguranță a reziduurilor.
20. Scara de măsurare, monitorizare sau afișare (inclusiv modificarea culorilor și a altor indicatori vizuali) trebuie să fie proiectată și fabricată în conformitate cu principiile ergonomice, conform scopului propus al dispozitivului.
22. Determinările realizate de dispozitivul cu funcție de măsurare vor fi exprimate în unități de măsură legale.
24. Dacă dispozitivele sînt destinate să emită radiații potențial periculoase, vizibile și/sau invizibile, ele necesită să fie pe cît posibil:
1) proiectate și realizate astfel încît să asigure că parametrii și cantitatea radiației emise pot fi controlate și/sau ajustate;
2) prevăzute cu afișaj vizual și/sau avertizare sonoră a acestor emisii.
25. Instrucțiunile de operare pentru dispozitivele ce emit radiații trebuie să ofere informații asupra naturii radiației emise, a mijloacelor de protecție a utilizatorului și a măsurilor ce pot fi întreprinse pentru evitarea utilizării necorespunzătoare și pentru eliminarea riscului inerent la instalare.
27. Dispozitivele trebuie să fie proiectate și realizate astfel încît să reducă pe cît posibil riscurile de generare a perturbațiilor electromagnetice, care ar putea afecta funcționarea altor dispozitive sau echipamente din mediul înconjurător uzual.
28. Dispozitivele trebuie să fie proiectate și fabricate astfel încît să evite pe cît posibil riscul șocului electric accidental în timpul folosirii normale și în condiții de prim defect, atunci cînd dispozitivele sînt instalate și întreținute corect.
30. Dispozitivele trebuie să fie proiectate și fabricate astfel încît să reducă la minimum riscurile ce apar din vibrația generată de dispozitive, bazîndu-se pe progresele tehnice și pe mijloacele disponibile pentru reducerea vibrației, în special la sursa de alimentare, numai dacă obținerea vibrațiilor nu reprezintă scopul propus.
31. Dispozitivele trebuie să fie proiectate și fabricate astfel încît să reducă cît se poate de mult riscurile care apar din zgomotul emis, în special la sursa de alimentare, dacă acest zgomot nu este parte a scopului propus.
32. Terminalele și conectorii alimentați de la o sursă de electricitate, de gaz, hidraulică sau pneumatică și care trebuie să fie mînuiți de utilizator trebuie să fie proiectați astfel încît să minimizeze toate riscurile posibile.
33. Părțile accesibile ale dispozitivelor (cu excepția părților sau a suprafețelor proiectate pentru a emite căldură sau pentru a atinge temperaturi prestabilite) și împrejurimile acestora nu trebuie să atingă temperaturile potențial periculoase în condiții de utilizare normală.
35. Dispozitivele pentru autotestare trebuie să fie proiectate și realizate astfel încît:
1) să asigure utilizarea ușoară a dispozitivului de către utilizator pe parcursul tuturor etapelor procedurii;
2) să reducă pe cît posibil riscul erorilor de manevrare a dispozitivului și de interpretare a rezultatelor.
36. Dispozitivele pentru autotestare urmează să prevadă controlul utilizatorului, cum ar fi procedura prin care utilizatorul verifică în timpul utilizării dacă produsul îndeplinește scopul prevăzut.
Aceste informații trebuie să fie pe etichetă și în instrucțiunile de utilizare.
În măsura în care este posibil și potrivit, informațiile necesare pentru a folosi dispozitivele în siguranță trebuie să fie afișate chiar pe dispozitiv și/sau pe ambalajul în care sînt comercializate. Dacă nu se practică ambalarea individuală, informațiile se redactează într-o broșură furnizată cu unul sau mai multe dispozitive.
Instrucțiunile de folosire trebuie să fie incluse în ambalaj pentru unul sau mai multe dispozitive. În cazuri excepționale, bine justificate, instrucțiunile de folosire nu sînt necesare dacă dispozitivul va fi folosit corect și în siguranță fără aceste instrucțiuni.
38. Dacă este posibil, aceste informații trebuie să ia forma simbolurilor. Orice simbol/culoare de identificare trebuie să fie conform/conformă standardelor. În cazul în care nu există standarde, simbolurile și culorile trebuie să fie descrise în documentația furnizată odată cu dispozitivul.
39. În cazul dispozitivelor care conțin un preparat ce se consideră periculos, ținîndu-se seama de natura și cantitatea constituenților săi și de forma sub care aceștia se prezintă, se aplică cerințele de etichetare și simbolurile de pericol din legislația cu privire la substanțele și preparatele chimice periculoase. Dacă spațiul este insuficient pentru ca toate informațiile să fie plasate pe dispozitiv sau pe etichetă, simbolurile de pericol trebuie să fie plasate pe etichetă, iar celelalte informații se vor conține în instrucțiunile de utilizare.
40. Eticheta trebuie să conțină următoarele date:
1) denumirea și adresa producătorului. Pentru dispozitivele importate în scopul de a fi distribuite în Republica Moldova, eticheta sau ambalajul exterior sau instrucțiunile de folosire vor conține în plus numele și adresa reprezentantului autorizat al producătorului;
2) detaliile strict necesare utilizatorului pentru identificarea fără posibilitate de eroare, a dispozitivului și a conținutului pachetului;
3) cuvîntul „STERIL” sau inscripția cu referire la starea microbiologică specială sau starea de puritate, acolo unde este cazul;
4) numărul lotului precedat de cuvîntul „LOT” sau numărul de serie, după caz;
5) dacă este necesar, data pînă la care dispozitivul poate fi utilizat în siguranță, fără afectarea performanței, exprimată, în ordine, prin anul, luna și, după caz, ziua;
6) în cazul dispozitivelor pentru evaluarea performanței, inscripția „pentru evaluarea performanței”;
7) inscripția că dispozitivul este utilizat in vitro;
8) condițiile speciale de păstrare și/sau de manevrare;
9) instrucțiunile speciale de utilizare;
10) atenționările și/sau precauțiile necesare;
11) dacă dispozitivul este destinat autotestării, precizarea clară a acestui fapt.
41. Dacă scopul propus al dispozitivului nu este evident pentru utilizator, producătorul trebuie să definească, în mod clar, în instrucțiunile de utilizare și, dacă este cazul, pe etichetă, domeniul și funcțiile dispozitivului.
42. Acolo unde este posibil, dispozitivul și părțile sale detașabile trebuie să fie accesibile pentru a fi identificate în numărul de lot și pentru a face posibilă detectarea oricărui risc potențial al dispozitivului și părților sale componente.
43. Instrucțiunile de utilizare trebuie să conțină următoarele:
1) datele prevăzute la pct. 40 din prezenta anexă, cu excepția subpct. 4) și 5);
2) compoziția reactivului, prin natura și cantitatea sau concentrația ingredientelor active ale reactivului (reactivilor) sau kitului (trusei), precum și precizarea, dacă este cazul, că dispozitivul conține alte ingrediente ce ar putea afecta măsurarea;
3) condițiile de depozitare și durata de păstrare după prima deschidere a containerului primar, împreună cu condițiile de depozitare și stabilitate a reactivilor de lucru;
4) performanțele menționate la pct. 3 din prezenta anexă;
5) precizarea oricărui echipament special necesar, incluzînd informații pentru identificarea acelui echipament în vederea utilizării corecte;
6) tipul de probe utilizate, condițiile speciale de recoltare, pretratament și, dacă este necesar, condițiile de depozitare și instrucțiunile pentru pregătirea pacientului;
7) descrierea detaliată a procedurii de urmat la utilizarea dispozitivului;
8) procedura de măsurare care trebuie respectată la utilizarea dispozitivului, inclusiv, dacă este cazul:
a) principiul care stă la baza metodei;
b) caracteristicile de performanță analitică specifice (de exemplu: sensibilitate, specificitate, acuratețe, repetabilitate, reproductibilitate, limite de detectare și intervalul de măsurare, inclusiv informații necesare pentru controlul unor interferențe importante cunoscute), limite ale metodei și informații despre folosirea de către utilizator a unor materiale și proceduri de referință disponibile;
c) detalii asupra oricărei alte proceduri suplimentare sau manipulări necesare înainte de utilizarea dispozitivului (de exemplu: reconstituire, incubare, diluare, verificarea instrumentelor);
d) indicația dacă este necesară o pregătire specială;
9) expresia matematică în baza căreia se calculează rezultatul analitic;
10) măsurările ce trebuie efectuate în cazul modificării performanței analitice a dispozitivului;
11) informațiile necesare utilizatorilor cu privire la:
a) controlul intern al calității, inclusiv procedurile de validare specifice;
b) calibrarea;
12) intervalele de referință pentru cantitățile ce sînt determinate, incluzînd descrierea populației de referință corespunzătoare;
13) dacă dispozitivul trebuie să fie instalat sau conectat la alte dispozitive medicale pentru a funcționa conform scopului propus, detaliile cu privire la caracteristicile sale pentru identificarea dispozitivelor ce necesită să fie utilizate pentru a se obține un sistem corect și sigur;
14) toate informațiile necesare pentru verificarea instalării corespunzătoare a dispozitivului, a utilizării sale corecte și sigure, detaliile asupra naturii și frecvenței operațiunilor de întreținere și calibrare, necesare în vederea asigurării utilizării corecte și sigure pe toată durata de funcționare; informațiile privind distrugerea în siguranță a reziduurilor;
15) detaliile pentru orice tratament sau manevră necesară înainte ca dispozitivul să fie utilizat (de exemplu, sterilizare, asamblare finală);
16) instrucțiunile necesare în caz de deteriorare a ambalajului protector și, dacă este cazul, metoda potrivită de resterilizare sau decontaminare;
17) dacă dispozitivul este reutilizabil, indicațiile privind procedurile necesare pentru reutilizare, inclusiv pentru curățare, dezinfectare, ambalare, resterilizare sau decontaminare, precum și orice restricții privind numărul de reutilizări;
18) precauțiile privind expunerea, în condiții de mediu previzibile, la cîmpuri magnetice, influențe electrice externe, descărcări electrostatice, presiune sau variație de presiune, accelerație și surse de aprindere termică;
19) precauțiile privind orice risc special, neobișnuit legat de utilizarea sau de distrugerea dispozitivului, inclusiv măsuri de protecție speciale. Dacă dispozitivul include substanțe de origine umană sau animală, trebuie avută în vedere natura lor potențial infecțioasă;
20) specificațiile pentru dispozitivele de autotestare:
a) rezultatele trebuie să fie exprimate și prezentate astfel încît să fie înțelese ușor de un neprofesionist; informațiile trebuie să fie însoțite de îndrumări pentru utilizator asupra modului de a acționa (în cazul unui rezultat pozitiv, negativ sau neconcludent) și asupra posibilității unor rezultate fals pozitive sau fals negative;
b) anumite particularități pot fi omise cu condiția ca alte informații furnizate de producător să fie suficiente pentru a permite utilizatorului să folosească dispozitivul și să înțeleagă rezultatele oferite de dispozitiv;
c) informația furnizată trebuie să includă un enunț care să îl determine pe utilizator să nu ia nicio decizie medicală relevantă înainte de a-și consulta medicul curant;
d) informația trebuie să precizeze de asemenea că, în cazul în care dispozitivul destinat autotestării este utilizat pentru monitorizarea unei afecțiuni existente, pacientul trebuie doar să-și adapteze tratamentul, dacă a fost pregătit corespunzător în acest sens;
21) data apariției sau a ultimei revizuiri a instrucțiunilor de utilizare.
2) reactivi și produși de reacție, inclusiv calibratori și materiale de referință pentru detectarea, confirmarea și cuantificarea în probele prelevate din organismul uman a markerilor infecției cu HIV (HIV 1 și HIV 2), HTLV I și II și virus hepatic B, C și D;
3) teste Varianta bolii Creutzfeldt-Jakob (vCJD) pentru screeningul sîngelui, diagnostic și confirmare.
2) reactivi și produși de reacție, inclusiv calibratori și materiale de referință pentru determinarea anticorpilor atipici antieritrocitari;
3) reactivi și produși de reacție, inclusiv calibratori și materiale de referință pentru detectarea și cuantificarea în probele prelevate din organismul uman a următoarelor infecții congenitale: rubeola și toxoplasmoza;
4) reactivi și produși de reacție, inclusiv calibratori și materiale de referință pentru diagnosticarea bolii ereditare fenilcetonurie;
5) reactivi și produși de reacție, inclusiv calibratori și materiale de referință pentru determinarea următoarelor infecții la om: citomegalovirus, chlamydia;
6) reactivi și produși de reacție, inclusiv calibratori și materiale de referință pentru determinarea următoarelor grupuri tisulare HLA: DR, A și B;
7) reactivi și produși de reacție, inclusiv calibratori și materiale de referință pentru determinarea markerilor tumorali PSA;
8) reactivi și produși de reacție, inclusiv calibratori, materiale de referință și software, destinate în special evaluării riscului de trisomie 21;
9) dispozitive pentru autodiagnostic, inclusiv, calibratori și materiale de referință pentru măsurarea nivelului de glucide în sînge.
2. Producătorul trebuie să pregătească documentația tehnică descrisă la pct. 3 din prezenta anexă și să asigure că în procesul de fabricație sînt respectate principiile de asigurare a calității stabilite la pct. 4 din prezenta anexă.
3. Documentația tehnică trebuie să permită evaluarea conformității produsului cu cerințele prezentului Regulament.
Aceasta trebuie să conțină, în special:
1) descrierea generală a produsului, incluzînd toate variantele proiectate;
2) documentația asupra sistemului calității;
3) informația privind proiectarea, inclusiv determinarea caracteristicilor materialelor de bază, a caracteristicilor și limitelor performanței dispozitivelor, metodele de fabricație și, în cazul instrumentelor, schițele de proiect, diagramele componentelor, subansamblurile, circuitele etc.;
4) în cazul dispozitivelor ce conțin țesuturi de origine umană sau substanțe derivate din astfel de țesuturi, informația privind originea acestora și condițiile în care au fost recoltate;
5) descrierile și explicațiile necesare pentru a înțelege caracteristicile, schițele și diagramele menționate mai sus și modul de utilizare a produsului;
6) rezultatele analizei riscurilor și, după caz, lista standardelor menționate în pct. 18 din prezentul Regulament, aplicate în întregime sau în parte, și descrierile soluțiilor adoptate pentru a îndeplini cerințele esențiale ale prezentului Regulament, dacă standardele menționate la pct. 18 nu au fost aplicate în întregime;
7) în cazul produselor sterile sau într-o stare microbiologică de curățenie specială, descrierea procedurilor utilizate;
8) rezultatele calculelor de proiectare și ale inspecțiilor efectuate;
9) dacă dispozitivul urmează să fie asociat cu alte dispozitive pentru a funcționa conform scopului propus, este necesar să fie aduse dovezi că dispozitivul satisface cerințele esențiale atunci cînd este asociat cu oricare dintre aceste dispozitive avînd caracteristicile specificate de producător;
10) rapoartele de încercări;
11) datele adecvate privind evaluarea performanței, care să indice performanțele prevăzute de producător și susținute de un sistem de măsurare de referință (atunci cînd este disponibil), cu informații despre metodele de referință, materialele de referință, valorile de referință cunoscute, precizia și unitățile de măsură utilizate; datele urmează să provină din studiile făcute în condiții clinice sau în alt mediu adecvat ori din referințe biografice relevante;
12) etichetele și instrucțiunile de utilizare;
13) rezultatele studiilor de stabilitate.
4. Producătorul ia măsurile necesare pentru a se asigura că în procesul de fabricație se respectă principiile de asigurare a calității adecvate a produselor fabricate.
Sistemul calității se referă la:
1) structurile organizatorice și responsabilități;
2) procesele de fabricație și controlul sistematic al calității producției;
3) mijloacele de monitorizare a performanțelor sistemului calității.
5. Producătorul instituie și actualizează permanent o procedură sistematică de analiză a experienței obținute în ceea ce privește dispozitivele în faza de postproducție și de aplicare a acțiunilor corective necesare, ținînd cont de natura și de riscurile legate de produs. Acesta trebuie să comunice Agenției următoarele incidente imediat ce a luat cunoștință de ele. La acestea se raportă:
1) orice disfuncție, defectare sau deteriorare a caracteristicilor și/sau a performanțelor dispozitivului, precum și orice neconformitate în etichetare sau în instrucțiunile de utilizare, care, direct ori indirect, ar putea sau ar fi putut conduce la decesul unui pacient, al unui utilizator ori al unei alte persoane sau la deteriorarea severă a stării de sănătate a acestora;
2) orice cauză de ordin tehnic sau medical legată de caracteristicile sau de performanța dispozitivului, care, pentru motivele menționate la subpct. 1) din prezentul punct, conduce la retragerea sistematică a dispozitivelor de același tip de către producător.
6. Pentru dispozitivele destinate autotestării, producătorul trebuie să depună o cerere în scopul examinării proiectului la un organism notificat, ales în conformitate cu prevederile prezentului Regulament.
7. Cererea trebuie să permită înțelegerea proiectului dispozitivului și evaluarea conformității cu cerințele prezentului Regulament aplicabile proiectului.
Ea va include:
1) rapoartele de testare, incluzînd, după caz, rezultatele studiilor realizate cu neprofesioniști;
2) datele ce relevă posibilitatea de manipulare a dispozitivului, ținînd cont de scopul propus pentru autotestare;
3) informațiile ce vor fi furnizate împreună cu dispozitivul pe etichetă și în instrucțiunile de utilizare.
8. Organismul notificat examinează cererea și, dacă proiectul este în conformitate cu prevederile prezentului Regulament, eliberează solicitantului un certificat de examinare a proiectului. Organismul notificat poate cere ca documentația să fie completată cu probe și teste ulterioare, care să permită evaluarea conformității cu cerințele prezentului Regulament aplicabile proiectului. Certificatul conține concluziile examinării, condițiile de validitate, datele necesare pentru identificarea proiectului aprobat și, după caz, descrierea scopului propus al dispozitivului.
9. Solicitantul informează organismul notificat care a eliberat certificatul de examinare a proiectului despre orice modificare semnificativă a proiectului aprobat. Modificările din proiectul aprobat trebuie să fie aprobate ulterior de organismul notificat care a eliberat certificatul de examinare a proiectului, dacă schimbările ar putea afecta conformitatea cu cerințele esențiale ale prezentului Regulament sau cu condițiile prevăzute pentru utilizarea produsului. Această aprobare suplimentară va lua forma unui document adițional la certificatul de examinare a proiectului.
2. Declarația de conformitate este procedura prin care producătorul care îndeplinește cerințele impuse la pct. 1 din prezenta anexă asigură și declară că dispozitivele îndeplinesc prevederile prezentului Regulament ce le sînt aplicabile.
Producătorul aplică marcajul în conformitate cu prevederile prezentului Regulament și emite declarația de conformitate referitoare la dispozitivele examinate.
3. Cerințele sistemului calității sînt următoarele:
1) producătorul trebuie să depună la un organism notificat o cerere pentru atestarea sistemului calității, care va include:
a) denumirea sau numele, după caz, și adresa producătorului și ale oricărui loc de fabricație în care se asigură sistemul calității;
b) informațiile relevante cu privire la dispozitiv sau la categoria de dispozitive acoperite de procedură;
c) declarația scrisă în care se menționează că nu a fost depusă o cerere la un alt organism notificat pentru același dispozitiv cu privire la sistemul calității;
d) documentația privind sistemul calității;
e) angajamentul producătorului de a acoperi în totalitate cerințele impuse prin sistemul calității aprobat;
f) angajamentul producătorului de a menține în mod corespunzător sistemul calității aprobat;
g) angajamentul producătorului de a institui și de a ține la zi o procedură sistematică de analiză a experienței obținute cu dispozitivele în faza de postproducție și de a implementa mijloacele adecvate pentru a aplica orice acțiune corectivă necesară sau notificare, după cum se precizează la pct. 5 din anexa nr. 3 la prezentul Regulament.
Sistemul calității trebuie să se refere la:
- structurile organizatorice și responsabilități;
- procesele de fabricație și controlul sistematic al calității producției;
- mijloacele de monitorizare a performanțelor sistemului calității;
2) aplicarea sistemului calității trebuie să asigure că dispozitivele sînt conforme cu prevederile prezentului Regulament ce le sînt aplicabile, la toate etapele, de la proiectare pînă la inspecția finală. Toate elementele, cerințele și prevederile adoptate de producător pentru sistemul său de calitate trebuie să fie documentate în mod sistematic și ordonat sub formă de proceduri și declarații scrise privind politicile de calitate, cum ar fi planuri, programe și înregistrări de calitate.
Acestea trebuie să cuprindă, în special, o descriere adecvată:
a) a obiectivelor producătorului privind calitatea;
b) a modului de organizare a producerii și, în particular:
- structurile organizatorice, responsabilitățile echipei de conducere și a autorității organizatorice în ceea ce privește calitatea proiectării și fabricării dispozitivelor;
- metodele de monitorizare a eficienței sistemului calității și, în particular, capacitatea lui de a atinge calitatea dorită în proiectare și în producere, inclusiv controlul dispozitivelor ce nu se conformează cerințelor de calitate;
c) a procedurilor de monitorizare și de verificare a proiectului dispozitivului și, în special:
- descrierea generală a dispozitivului, inclusiv a tuturor variantelor planificate;
- documentația menționată la pct. 3 subpct. 3)-13) din anexa nr. 3 la prezentul Regulament;
- în cazul dispozitivelor destinate autotestării, informațiile menționate la pct. 6 și 7 din anexa nr. 3 la prezentul Regulament;
- tehnicile utilizate pentru controlul și verificarea proiectului și a proceselor și măsurilor sistematice ce vor fi utilizate în proiectare;
d) a tehnicilor de inspecție și de asigurare a calității în stadiul de producție și, în particular:
- procesele și procedurile care vor fi utilizate, în special cu privire la sterilizare;
- procedurile legate de achiziționare;
- procedurile de identificare a produsului, schițate și actualizate prin proiecte, specificații sau alte elemente relevante, în orice stadiu de producție;
e) a încercărilor și verificărilor adecvate, care trebuie să fie efectuate înainte, în timpul și după fabricație, a frecvenței cu care vor avea loc și a echipamentelor de testare utilizate; trebuie să fie asigurată trasabilitatea măsurărilor.
Producătorul trebuie să realizeze controalele și încercările cerute în conformitate cu nivelul actual de dezvoltare a tehnologiei în domeniu. Controalele și încercările trebuie să acopere procesul fabricației, inclusiv caracteristicile materiilor prime și dispozitivele individuale sau fiecare lot de dispozitive fabricate.
La testarea dispozitivelor prevăzute în lista A din anexa nr. 2 la prezentul Regulament, producătorul urmează să se bazeze pe cele mai recente informații disponibile, în particular în ceea ce privește complexitatea biologică și variabilitatea specimenelor de testat cu dispozitivul pentru diagnostic in vitro respectiv;
3) organismul notificat trebuie să auditeze sistemul calității pentru a stabili conformitatea cu cerințele menționate la subpct. 2) din prezentul punct. Se presupune că sistemul calității care implementează standardele relevante este conform cu aceste cerințe.
Echipa de evaluare trebuie să aibă, în mod obligatoriu, experiență în evaluarea tehnologiilor respective. Procedura de evaluare include inspecția la locul de producție și, în cazuri justificate, la locul de producție al furnizorilor și/sau al subcontractanților, pentru a inspecta procesul de fabricație.
Decizia inspecției, care conține o evaluare argumentată și concluziile inspecției, este comunicată producătorului;
4) producătorul informează organismul notificat care a aprobat sistemul calității despre orice plan de modificare substanțială a sistemului calității sau a gamei de produse acoperite de acest sistem.
Organismul notificat trebuie să evalueze modificările propuse și să verifice dacă după operarea acestor modificări sistemul calității își menține conformitatea cu cerințele menționate la subpct. 2) din prezentul punct. Rezultatele evaluării argumentate și decizia privind rezultatul inspecției, care conține concluziile inspecției, urmează să fie comunicate producătorului.
4. Cerințele pentru examinarea proiectului produsului sînt următoarele:
1) pentru dispozitivele prevăzute în lista A din anexa nr. 2 la prezentul Regulament, suplimentar la obligațiile impuse la pct. 3 din prezenta anexă, producătorul depune la organismul notificat o cerere de examinare a dosarului de proiectare a dispozitivului pe care intenționează să îl fabrice și care intră în categoria menționată la pct. 3 subpct.1) din prezenta anexă;
2) cererea trebuie să descrie proiectul, procesul de fabricație și performanțele dispozitivului și să includă documentele indicate în pct. 3 subpct. 2) lit. c) din prezenta anexă pentru a aprecia dacă dispozitivul este în conformitate cu cerințele prezentului Regulament;
3) organismul notificat examinează cererea și dacă dispozitivul corespunde prevederilor prezentului Regulament, eliberează certificatul de examinare a proiectului. Organismul notificat poate solicita ca cererea să fie completată cu încercări sau cu probe suplimentare care să permită evaluarea conformității cu cerințele Regulamentului.
Certificatul va conține concluziile examinării, condițiile de validitate, datele necesare pentru identificarea proiectului aprobat și, după caz, descrierea scopului propus al dispozitivului;
4) modificările operate în proiectul aprobat trebuie să obțină în prealabil aprobarea organismului notificat care a emis certificatul de examinare a proiectului, dacă aceste modificări ar putea afecta conformitatea cu cerințele esențiale sau cu condițiile prevăzute pentru utilizarea dispozitivului. Solicitantul trebuie să informeze organismul notificat care a emis certificatul de examinare a proiectului despre orice schimbare efectuată în proiectul aprobat. Aprobarea adițională constituie un supliment la certificatul de examinare a proiectului;
5) producătorul este obligat să comunice fără întîrziere organismului notificat dacă a obținut informații despre orice schimbare a agenților patogeni sau a markerelor de infecție ce vor fi testați, în particular ca o consecință a variabilității și complexității biologice. În acest sens, producătorul va informa organismul notificat dacă o astfel de modificare ar putea afecta performanțele respectivului dispozitiv medical pentru diagnostic in vitro.
5. Supravegherea urmează să satisfacă următoarele cerințe:
1) scopul supravegherii este de a garanta că producătorul îndeplinește obligațiile impuse prin sistemul calității aprobat;
2) producătorul permite organismului notificat să efectueze inspecțiile necesare și îi furnizează orice informații relevante în legătură cu:
a) documentația privind sistemul calității;
b) datele prevăzute în sistemul calității cu privire la proiect, cum ar fi rezultatele analizelor, calculelor, încercărilor etc.;
c) datele prevăzute în sistemul calității cu privire la producție, cum ar fi rapoartele de inspecție, datele de testare și calibrare, rapoartele privind calificarea personalului;
3) organismul notificat efectuează periodic inspecții și evaluări pentru a se asigura că producătorul aplică sistemul calității aprobat și îi transmite producătorului un raport de inspecție;
4) suplimentar, organismul notificat face vizite inopinate producătorului, în cadrul cărora cere să se efectueze încercări pentru verificarea aplicării corecte a sistemului calității. Acesta furnizează producătorului un raport de inspecție sau de încercări, după caz.
6. Verificarea produselor fabricate prevăzute în lista A din anexa nr. 2 la prezentul Regulament se efectuează în felul următor:
1) în cazul dispozitivelor prevăzute în lista A din anexa nr. 2, producătorul trebuie să înainteze fără întîrziere organismului notificat, după încheierea controalelor și testelor, rapoartele asupra testelor realizate pe dispozitivele fabricate sau pe fiecare lot de dispozitive. Suplimentar, producătorul pune la dispoziția organismului notificat mostre ale dispozitivelor sau ale loturilor de dispozitive fabricate în conformitate cu condițiile și modalitățile prestabilite;
2) producătorul are dreptul să introducă dispozitivele pe piață dacă organismul notificat nu îi comunică în termenul prestabilit, dar nu mai tîrziu de expirarea a 30 de zile de la data primirii mostrelor, o altă decizie, incluzînd în particular orice condiție de validitate a certificatelor emise.
2. Cererea pentru examinarea de tip va fi depusă de producător ori de reprezentantul său autorizat la un organism notificat și include:
1) numele și adresa producătorului și ale reprezentantului autorizat, dacă cererea este depusă de acesta din urmă;
2) documentația menționată la pct. 3 din prezenta anexă, necesară pentru evaluarea conformității mostrei reprezentative (în continuare – tip) cu cerințele prezentului Regulament. Solicitantul trebuie să prezinte un astfel de tip organismului notificat, iar acesta cere și alte mostre, după necesitate;
3) declarația scrisă prin care se menționează că examinarea aceluiași tip nu a fost solicitată altui organism notificat.
3. Documentația trebuie să permită înțelegerea proiectului, a procesului de fabricare și a performanțelor dispozitivului și să cuprindă în special următoarele aspecte:
1) descrierea generală a tipului, inclusiv toate variantele planificate;
2) toată documentația prevăzută la pct. 3 subpct. 3)-13) din anexa nr. 3 la prezentul Regulament;
3) în cazul dispozitivelor pentru autotestare, informațiile menționate la pct. 6 și 7 din anexa nr. 3 la prezentul Regulament.
4. Organismul notificat trebuie:
1) să examineze și să aprobe documentația, să verifice dacă tipul a fost fabricat în conformitate cu această documentație; să înregistreze produsele proiectate în conformitate cu prevederile aplicabile ale standardelor la care se referă pct. 18 din prezentul Regulament, precum și produsele care nu au fost proiectate conform acestor standarde;
2) să efectueze sau să organizeze inspecțiile și încercările necesare pentru a verifica soluțiile adoptate de producător și dacă respectă cerințele esențiale din prezentul Regulament, în cazul în care standardele la care se referă pct. 18 din prezentul Regulament nu au fost aplicate. Dacă dispozitivul este destinat să fie asociat cu alte dispozitive pentru a fi utilizat conform scopului propus, trebuie aduse dovezi că acesta corespunde cerințelor esențiale atunci cînd este asociat cu astfel de dispozitive avînd caracteristicile specificate de către producător;
3) să efectueze sau să solicite inspecțiile și încercările necesare pentru a verifica, în cazul în care producătorul a decis să aplice standardele relevante, dacă acestea au fost într-adevăr aplicate;
4) să stabilească, de comun acord cu solicitantul, locul unde vor fi efectuate inspecțiile și încercările necesare.
5. Dacă tipul este conform cu prevederile prezentului Regulament, organismul notificat emite un certificat de examinare CE de tip. Certificatul va conține numele și adresa producătorului, concluziile inspecției, condițiile de validitate și datele necesare pentru identificarea tipului aprobat. Documentația relevantă este anexată la certificat, iar o copie se păstrează la organismul notificat.
6. Producătorul este obligat să informeze fără întîrziere organismul notificat dacă a obținut informații despre orice modificări ale agenților patogeni și ale markerelor infecțiilor ce urmează a fi testați, în particular ca o consecință a variabilității și complexității biologice. În acest sens, producătorul va informa organismul notificat dacă o astfel de modificare ar putea afecta performanțele respectivului dispozitiv medical pentru diagnostic in vitro.
7. Modificările dispozitivului aprobat trebuie să obțină aprobarea suplimentară a organismului notificat care a emis certificatul de examinare are CE de tip, dacă aceste modificări afectează conformitatea cu cerințele esențiale sau cu condițiile de utilizare prescrise. Solicitantul va informa organismul notificat care a emis certificatul de examinare de tip despre orice modificare a dispozitivului aprobat. Noua aprobare se emite sub forma unui supliment la certificatul inițial de examinare CE de tip.
8. Alte organisme notificate pot obține o copie de pe certificatul de examinare CE de tip și/sau de pe suplimentele acestuia. Anexele la certificat trebuie să fie accesibile și altor organisme notificate, la solicitarea argumentată a acestora, după informarea prealabilă a producătorului.
2. Producătorul trebuie să ia toate măsurile necesare pentru a se asigura că din procesul de fabricație rezultă produse conforme cu tipul descris în certificatul de examinare de tip și cu cerințele prezentului Regulament. Înaintea începerii fabricației, producătorul trebuie să își întocmească documentele care definesc procesul de fabricație, în special privind sterilizarea și caracterul adecvat al materiilor prime, dacă este necesar, și să definească procedurile necesare de testare la nivelul tehnologiilor de vîrf. Toate prescripțiile de rutină prestabilite urmează să fie implementate pentru a se asigura omogenitatea producției și conformitatea produselor cu tipul descris în certificatul de examinare de tip și cu cerințele prezentului Regulament ce le sînt aplicabile.
În măsura în care pentru anumite aspecte testarea finală, conform pct. 6 subpct. 3) din prezenta anexă, nu este adecvată, testarea, monitorizarea și metodele de control adecvate ale proceselor de fabricație urmează să fie stabilite de către producător, cu aprobarea organismului notificat. Prevederile pct. 5 din anexa nr. 4 la prezentul Regulament se aplică corespunzător procedurilor sus-menționate.
3. Producătorul trebuie să instituie și să actualizeze permanent proceduri sistematice de valorificare a experienței obținute privind dispozitivele în faza de postproducție și să implementeze măsurile corespunzătoare pentru aplicarea oricărei acțiuni corective necesare și pentru notificare, după cum se menționează la pct. 5 din anexa nr. 3 la prezentul Regulament.
4. Organismul notificat trebuie să efectueze examinările și încercările necesare ținînd seama de pct. 2 din prezenta anexă, pentru verificarea conformității produsului cu cerințele prezentului Regulament, fie prin examinarea și încercarea fiecărui produs, după cum se menționează la pct. 5 din prezenta anexă, fie prin examinarea și încercarea statistică, conform pct. 6 din prezenta anexă, la decizia producătorului. La realizarea verificării statistice conform pct. 6, organismul notificat decide cînd trebuie aplicate procedurile statistice pentru inspectarea fiecărui lot în parte sau pentru inspectarea de loturi izolate. O astfel de decizie se ia prin consultarea cu producătorul.
Dacă efectuarea examinărilor și încercărilor pe baze statistice nu este adecvată, examinările și încercările se pot face în mod aleatoriu, cu condiția ca o astfel de procedură asociată cu măsurile adoptate în conformitate cu pct. 2 alineatul doi din prezenta anexă să asigure un nivel echivalent de conformitate.
5. Verificarea prin examinarea și încercarea fiecărui produs se efectuează respectîndu-se următoarele cerințe:
1) fiecare produs este examinat individual și se efectuează încercările necesare definite în standardele relevante menționate în pct. 18 din prezentul Regulament sau alte încercări echivalente pentru verificarea conformității produsului cu tipul descris în certificatul de examinare de tip și cu cerințele prezentului Regulament;
2) organismul notificat trebuie să aplice sau să asigure că se aplică numărul său de identificare pe fiecare produs aprobat și să emită în scris un certificat de conformitate privind încercările efectuate.
6. Cerințele față de verificările statistice sînt următoarele:
1) producătorul trebuie să prezinte produsele fabricate sub formă de loturi omogene;
2) se iau, la întîmplare, una sau mai multe mostre din fiecare lot. Produsele care alcătuiesc mostra sînt examinate conform standardelor relevante și se efectuează testările adecvate, definite în standardele menționate la pct. 18 din prezentul Regulament, sau încercările echivalente pentru verificarea conformității produselor cu tipul descris în certificatul de examinare de tip și cu cerințele prezentului Regulament, pentru a stabili dacă lotul se acceptă sau se respinge;
3) procedura de control statistic al produselor se bazează pe atribute și/sau variabile, necesitînd metode de prelevare a mostrelor cu caracteristici operaționale care să asigure un grad înalt de siguranță și performanță, ținînd cont de nivelul actual de dezvoltare a tehnologiei de vîrf în domeniu. Metoda de prelevare a mostrelor este stabilită prin standardele menționate în pct. 18 din prezentul Regulament, ținîndu-se cont de natura specifică a categoriilor de produse respective;
4) dacă lotul este acceptat, organismul notificat aplică sau se asigură că se aplică numărul său de identificare pe fiecare produs și eliberează un certificat de conformitate privind încercările efectuate. Toate produsele unui lot sînt introduse pe piață, cu excepția cazului în care mostrele au fost necorespunzătoare;
5) dacă un lot este respins, organismul notificat trebuie să ia măsurile necesare pentru a preveni introducerea pe piață a lotului respectiv. În eventualitatea respingerii frecvente a loturilor, organismul notificat suspendă verificarea statistică;
6) producătorul este în drept, sub responsabilitatea organismului notificat, să aplice numărul de identificare al acestuia în timpul procesului de fabricație.
2. Declarația de conformitate este parte a procedurii prin care producătorul care îndeplinește obligațiile impuse la pct. 1 din prezenta anexă asigură și declară că produsele respective sînt conforme cu tipul descris în certificatul de examinare de tip și corespund dispozițiilor aplicabile ale prezentului Regulament.
Producătorul trebuie să aplice marcajul CE în conformitate cu prevederile prezentului Regulament și să întocmească o declarație scrisă de conformitate cu privire la dispozitivele respective.
3. Sistemul calității este organizat în felul următor:
1) producătorul trebuie să depună la un organism notificat o cerere pentru evaluarea sistemului calității, care va cuprinde:
a) toate documentele și mențiunile specificate la pct. 3 subpct.1) din anexa nr. 4 la prezentul Regulament;
b) documentația tehnică a tipurilor aprobate și copia de pe certificatul de examinare CE de tip;
2) aplicarea sistemului calității trebuie să asigure că dispozitivele sînt conforme cu tipul descris în certificatul de examinare CE de tip.
Toate elementele, cerințele și prevederile adoptate de producător pentru sistemul său de calitate trebuie să fie documentate în mod sistematic și ordonat sub formă de proceduri și declarații scrise privind politica de calitate. Documentația sistemului de calitate trebuie să permită interpretarea uniformă a politicii de calitate și a procedurilor, cum ar fi planuri, programe, manuale și înregistrări de calitate.
Documentația trebuie să cuprindă, în special, o descriere adecvată:
a) a obiectivelor producătorului privind sistemul calității;
b) a modului de organizare a activității și, în particular:
- structurile organizatorice, responsabilitățile echipei de conducere și a autorității organizatorice în ceea ce privește calitatea proiectării și fabricării dispozitivelor;
- metodele de monitorizare a eficienței sistemului calității și, în particular, capacitatea lui de a atinge calitatea dorită în proiectare și în producere, inclusiv controlul dispozitivelor ce nu se conformează cerințelor;
c) a tehnicilor de inspecție și de asigurare a calității în stadiul de producție, în particular:
- procesele și procedurile care vor fi utilizate, în special cu privire la sterilizare;
- procedurile legate de achiziționare;
- procedurile de identificare a produsului, schițate și actualizate prin proiecte, specificații sau alte elemente relevante, în orice stadiu de producție;
d) a încercărilor și verificărilor care trebuie să fie efectuate înainte, în timpul și după fabricație, a frecvenței cu care vor avea loc și a echipamentelor de testare utilizate; trebuie să fie asigurată trasabilitatea măsurărilor;
3) organismul notificat trebuie să verifice sistemul calității pentru a determina dacă aceasta satisface cerințele menționate la pct. 3 subpct. 2) din prezenta anexă. Se presupune că sistemul de calitate care implementează standardele relevante este conform cu aceste cerințe.
Echipa de evaluare trebuie să aibă, în mod obligatoriu, experiență în evaluarea tehnologiilor respective. Procedura de evaluare trebuie să includă o inspecție la locul de producție și, în cazuri justificate, la locul de producție al furnizorilor și/sau al subcontractanților, pentru a inspecta procesul de fabricație.
Decizia inspecției, care conține o evaluare argumentată și concluziile acestuia, este comunicată producătorului;
4) producătorul informează organismul notificat care a aprobat sistemul calității asupra oricărui plan de modificare substanțială a sistemului calității.
Organismul notificat trebuie să evalueze modificările propuse și să verifice dacă după operarea acestor modificări sistemul calității își menține conformitatea cu cerințele menționate la pct. 3 subpct. 2) din prezenta anexă. Decizia privind rezultatul inspecției care conține concluziile inspecției și o evaluare argumentată, este comunicată producătorului;
5) supravegherea se aplică conform prevederilor pct. 5 din anexa nr. 4 la prezentul Regulament.
4. Verificarea produselor prevăzute în lista A din anexa nr. 2 la prezentul Regulament se realizează după cum urmează:
1) producătorul înaintează, fără întîrziere, organismului notificat, ținînd cont de concluziile controalelor și testelor, rapoartele asupra testelor realizate pe dispozitivele fabricate sau pe fiecare lot de dispozitive. Suplimentar, producătorul pune la dispoziția organismului notificat mostre de dispozitive sau ale loturilor de dispozitive fabricate în conformitate cu condițiile și modalitățile prestabilite;
2) producătorul poate introduce dispozitivele pe piață dacă organismul notificat nu îi comunică în termenul prestabilit, dar nu mai tîrziu de expirarea a 30 de zile de la recepționarea mostrelor, o altă decizie, incluzînd în particular orice condiție de validitate a certificatelor emise.
2. Declarația va conține următoarele informații:
1) datele ce permit identificarea dispozitivului respectiv;
2) planul de evaluare prin care se stabilesc scopul, bazele științifice, tehnice sau medicale, domeniul evaluării și numărul de dispozitive vizate;
3) lista laboratoarelor sau altor instituții care participă la studiul de evaluare;
4) data de pornire și durata programului de evaluare, iar în cazul dispozitivelor pentru autotestare, locul și numărul persoanelor neprofesioniste implicate;
5) declarația din care să rezulte că dispozitivul în cauză este conform cerințelor prezentului Regulament, cu excepția aspectelor acoperite de evaluare și a celor special menționate în declarație, și că au fost luate toate măsurile pentru protecția sănătății și siguranței pacientului, a utilizatorului și a altor persoane.
3. Producătorul trebuie să păstreze la dispoziția Agenției documentația care să permită înțelegerea proiectului, a procesului de fabricație și a performanțelor produsului, inclusiv a performanțelor preconizate, astfel încît să permită evaluarea conformității cu cerințele impuse de prezentul Regulament. Această documentație urmează să fie păstrată pentru o perioadă de cel puțin 5 ani de la data încheierii evaluării performanței.
Producătorul va lua toate măsurile necesare pentru asigurarea procesului de fabricație, astfel încît produsele să fie conforme cu documentația menționată în primul punct.
4. Prevederile prezentului Regulament se aplică și dispozitivelor destinate evaluării performanței.
Aceștia nu pot să fie direct implicați în proiectarea, construcția, introducerea pe piață sau întreținerea dispozitivelor și nici să reprezinte părțile angajate în astfel de activități. Aceasta însă nu exclude în niciun mod posibilitatea schimbului de informații tehnice între producător și organismul de evaluare a conformității notificat.
2. Organismul notificat și personalul său efectuează evaluarea și verificarea la cel mai înalt nivel de integritate profesională și competență în domeniul dispozitivelor medicale și este liber de orice presiune și influență, în special financiară, care le-ar putea influența decizia privind rezultatele inspecției, în special din partea persoanelor sau a grupurilor de persoane interesate de rezultatul verificărilor.
Dacă organismul de evaluare a conformității notificat subcontractează sarcini specifice în legătură cu stabilirea și verificarea faptelor, acesta se asigură mai întîi că subcontractantul respectă cerințele prezentului Regulament și, în special, pe cele ale prezentei anexe. Organismul de evaluare a conformității notificat păstrează la dispoziția Ministerul Sănătății, Muncii și Protecției Sociale documentele relevante de evaluare a calificărilor subcontractantului și cele privind activitatea acestuia care cad sub incidența prezentului Regulament.
3. Organismul de evaluare a conformității notificat este capabil să execute toate sarcinile atribuite acestor tipuri de organisme prin una dintre anexele nr. 3-7 la prezentul Regulament, sarcini pentru care a fost notificat, indiferent dacă aceste sarcini sînt îndeplinite de însuși organismul respectiv sau doar pe răspunderea lui. În special, organismul notificat dispune de personalul și de facilitățile necesare pentru îndeplinirea în mod corespunzător a sarcinilor tehnice și administrative aferente evaluării și verificării.
Acest lucru presupune existența în cadrul organizației a unui număr suficient de personal științific, care să posede experiență și cunoștințe suficiente pentru a evalua funcționalitatea medicală și performanța dispozitivelor pentru care a fost notificat, avînd în vedere cerințele prezentului Regulament, în special cele enunțate în anexa nr. 1 la Regulament.
Organismul de evaluare a conformității notificat trebuie, de asemenea, să aibă acces la echipamentul necesar pentru verificările cerute.
4. Personalul organismului de evaluare a conformității notificat are:
1) instruire profesională temeinică pentru operațiunile de evaluare și verificare pentru care organismul a fost notificat;
2) cunoștințe suficiente în domeniul reglementărilor cu privire la inspecțiile pe care le efectuează și o experiență adecvată;
3) capacitate adecvată pentru întocmirea certificatelor, a înregistrărilor și a rapoartelor ce demonstrează efectuarea inspecțiilor.
5. Imparțialitatea organismului de evaluare a conformității notificat este garantată. Salarizarea personalului acestuia nu depinde de numărul inspecțiilor efectuate și nici de rezultatele inspecțiilor.
6. Organismul de evaluare a conformității notificat are o asigurare de răspundere civilă, cu excepția cazului în care autoritatea competentă însăși efectuează inspecțiile în mod direct.
7. Personalul organismului de evaluare a conformității notificat este obligat să respecte secretul profesional cu privire la toate informațiile obținute în exercitarea atribuțiilor sale (cu excepția raporturilor cu autoritățile publice competente ale statului) conform prezentului Regulament sau oricărei prevederi legale în vigoare ce reglementează domeniul dispozitivelor medicale.
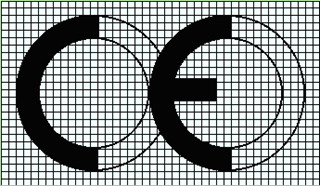
2. În cazul în care marcajul este micșorat sau mărit, trebuie respectate proporțiile date în desenul gradat de mai sus.
3. Diferitele componente ale marcajului CE au în principal aceeași dimensiune verticală, care nu poate fi mai mică de 5 mm.
4. La această dimensiune minimă se poate renunța în cazul dispozitivelor fabricate în serie mică.
1. Se aprobă Regulamentul privind condițiile de introducere pe piață a dispozitivelor medicale pentru diagnostic in vitro (se anexează)
2. Se admite introducerea pe piață și punerea în funcțiune a dispozitivelor medicale pentru diagnostic in vitro care poartă marcajul de conformitate SM, aplicat conform prevederilor din Legea nr. 235 din 1 decembrie 2011 privind activitățile de acreditare și de evaluare a conformității, și sînt însoțite de declarația de conformitate.
Producătorul sau reprezentantul autorizat al acestuia, persoană juridică cu sediul în Republica Moldova, aplică marcajul de conformitate SM în situația în care evaluarea conformității dispozitivelor medicale pentru diagnostic in vitro se realizează de către organismele de evaluare a conformității notificate prin utilizarea procedurilor prevăzute de Regulamentul privind condițiile de introducere pe piață a dispozitivelor medicale pentru diagnostic in vitro.
Se interzice, în condițiile prevăzute de Regulamentul menționat, aplicarea pe același dispozitiv a marcajului de conformitate SM și a marcajului CE.
3. Ministerul Sănătății, Muncii și Protecției Sociale recunoaște organismele acreditate conform prevederilor din Legea nr. 235 din 1 decembrie 2011 privind activitățile de acreditare și de evaluare a conformității, care realizează evaluarea conformității dispozitivelor medicale pentru diagnostic in vitro destinate pieței naționale în concordanță cu procedurile prevăzute în Regulamentul privind condițiile de introducere pe piață a dispozitivelor medicale pentru diagnostic in vitro.
4. Cerințele cu privire la organismele de evaluare a conformității notificate se aplică și organismelor de evaluare a conformității recunoscute. La desfășurarea procedurilor de evaluare a conformității, organismele de evaluare a conformității recunoscute care realizează evaluarea conformității dispozitivelor medicale pentru diagnostic in vitro vor întocmi certificate de examinare de tip.
5. Lista cuprinzînd organismele de evaluare a conformității recunoscute, sarcinile specifice pentru care acestea au fost recunoscute și numerele lor de identificare se publică în Monitorul Oficial al Republicii Moldova.
6. Obligațiile și răspunderea producătorului, reprezentantului său autorizat, ale importatorului sau distribuitorului, persoane juridice cu sediul în Republica Moldova, privind dispozitivele medicale pentru diagnostic in vitro puse în funcțiune care dețin marcajul de conformitate SM sînt similare cu cele prevăzute de prezenta hotărîre pentru dispozitivele medicale pentru diagnostic in vitro care dețin marcajul CE.
7. Toate activitățile ce țin de controlul de stat sau activitățile de supraveghere a pieței care înglobează în sine activitatea de control de stat, în special aplicarea măsurilor de interzicere sau restrîngere a plasării dispozitivelor medicale pentru diagnostic in vitro și activitățile ce țin de controlul de stat al dispozitivelor medicale pentru diagnostic in vitro de pe piață prevăzute de prezentul Regulament vor fi efectuate de către Agenția Națională pentru Sănătate Publică, în comun cu Agenția Medicamentului și Dispozitivelor Medicale, în conformitate cu prevederile Legii nr. 131 din 8 iunie 2012 privind controlul de stat asupra activității de întreprinzător și ale Legii nr.102 din 9 iunie 2017 cu privire la dispozitivele medicale.
8. Controlul asupra executării prezentei hotărîri se pune în sarcina Ministerului Sănătății, Muncii și Protecției Sociale.
9. Se abrogă Hotărîrea Guvernului nr. 435 din 10 iunie 2014 „Pentru aprobarea Regulamentului privind condițiile de plasare pe piață a dispozitivelor medicale pentru diagnostic in vitro” (Monitorul Oficial al Republicii Moldova, 2014, nr. 160-166, art. 482).
PRIM-MINISTRU Pavel FILIP
Contrasemnează:
Ministrul sănătății,
muncii și protecției sociale Svetlana Cebotari
Ministrul economiei și infrastructurii Chiril Gaburici
Ministrul afacerilor externe
și integrării europene Tudor Ulianovschi
Ministrul justiției Victoria Iftodi
Nr. 703. Chişinău, 11 iulie 2018.
Aprobat
prin Hotărîrea Guvernului nr. 703
din 11 iulie 2018
prin Hotărîrea Guvernului nr. 703
din 11 iulie 2018
REGULAMENT
privind condițiile de introducere pe piață a dispozitivelor
medicale pentru diagnostic in vitro Regulamentul privind condițiile de introducere
pe piață a dispozitivelor medicale pentru diagnostic in vitro (în continuare – Regulament)
transpune Directiva 98/79/CE a Parlamentului European și a Consiliului
din 27 octombrie 1998 privind dispozitivele medicale pentru diagnostic in vitro,
publicată în Jurnalul Oficial al Comunității Europene L 331 din 7 decembrie 1998.
Capitolul I
DEFINIȚII ȘI DOMENIUL DE APLICARE
1. Prezentul Regulament se aplică dispozitivelor medicale pentru diagnostic in vitro și accesoriilor acestora (în continuare – dispozitive). În sensul prezentului Regulament, accesoriile sînt tratate ca dispozitive medicale propriu-zise pentru diagnostic in vitro. privind condițiile de introducere pe piață a dispozitivelor
medicale pentru diagnostic in vitro Regulamentul privind condițiile de introducere
pe piață a dispozitivelor medicale pentru diagnostic in vitro (în continuare – Regulament)
transpune Directiva 98/79/CE a Parlamentului European și a Consiliului
din 27 octombrie 1998 privind dispozitivele medicale pentru diagnostic in vitro,
publicată în Jurnalul Oficial al Comunității Europene L 331 din 7 decembrie 1998.
Capitolul I
DEFINIȚII ȘI DOMENIUL DE APLICARE
2. În sensul prezentului Regulament se utilizează terminologia definită în Legea nr. 102 din 9 iunie 2017 cu privire la dispozitivele medicale, Legea nr. 235 din 1 decembrie 2011 privind activitățile de acreditare și de evaluare a conformității, Legea nr. 1409-XIII din 17 decembrie 1997 cu privire la medicamente, precum și următoarele noțiuni:
dispozitiv pentru autotestare – orice dispozitiv conceput de către producător astfel încât să fie folosit de nespecialiști în condiții casnice;
producător – persoană fizică sau juridică care are responsabilitatea de a proiecta, a produce, a ambala și a eticheta un dispozitiv medical pentru introducerea lui pe piață sub propria sa denumire, indiferent dacă această operațiune este efectuată de această persoană personal sau de o parte terță în numele ei (responsabilă de introducerea pe piață). Noțiunea se aplică și persoanelor fizice sau juridice care asamblează, ambalează, prelucrează, recondiționează și/sau etichetează produse și/sau atribuie acestora destinația de dispozitiv medical, cu intenția de introducere a acestuia pe piață sub propria sa denumire. Noțiunea nu se aplică persoanelor care, nefiind producători în înțelesul acestei definiții, asamblează sau adaptează dispozitive medicale deja existente pe piață pentru un anumit pacient;
dispozitiv de evaluare a performanței – dispozitiv destinat de către producător să fie supus unuia sau mai multor studii de evaluare a performanței în laboratoare pentru analize medicale sau într-un alt spațiu adecvat aflat în afara propriei sale incinte.
3. În sensul prezentului Regulament, materialele de control și etalon includ orice substanță, material sau articol destinat de producător fie să stabilească relații de măsurare, fie să calibreze sau să verifice caracteristicile de performanță ale unui dispozitiv în legătură cu scopul acestuia.
4. În sensul prezentului Regulament, îndepărtarea, recoltarea și utilizarea țesuturilor, a celulelor și a substanțelor de origine umană sînt reglementate prin Legea nr. 42-XVI din 6 martie 2008 privind transplantul de organe, țesuturi și celule umane.
5. Prevederile prezentului Regulament nu se aplică dispozitivelor fabricate și utilizate numai în cadrul aceleiași unități medicale și, respectiv, la sediul producătorului sau utilizate în clădiri din imediata vecinătate, fără să fi fost transferate unei alte persoane juridice. Aceasta nu va afecta dreptul Agenției Medicamentului și Dispozitivelor Medicale (în continuare – Agenție) de a supune astfel de activități unor cerințe specifice de protecție.
6. Prevederile prezentului Regulament nu aduc atingere actelor normative în vigoare privind furnizarea de dispozitive pe bază de prescripție medicală.
Capitolul II
INTRODUCEREA PE PIAȚĂ A DISPOZITIVELOR MEDICALE
PENTRU DIAGNOSTIC IN VITRO
Secțiunea 1. Cerințe esențiale de introducere pe piață
și punere în funcțiune a dispozitivelor medicale
7. Agenția ia toate măsurile necesare, în conformitate cu prevederile Legii nr. 102 din 9 iunie 2017 cu privire la dispozitivele medicale, pentru a se asigura că dispozitivele medicale pentru diagnostic in vitro pot fi introduse pe piață și/sau puse în funcțiune numai dacă se conformează prevederilor prezentului Regulament atunci cînd sînt furnizate și instalate corespunzător, întreținute și utilizate corect în conformitate cu scopul propus, inclusiv în ceea ce privește dispozitivele pentru evaluarea performanței.INTRODUCEREA PE PIAȚĂ A DISPOZITIVELOR MEDICALE
PENTRU DIAGNOSTIC IN VITRO
Secțiunea 1. Cerințe esențiale de introducere pe piață
și punere în funcțiune a dispozitivelor medicale
8. Dispozitivele trebuie să îndeplinească cerințele esențiale prevăzute în anexa nr. 1 la prezentul Regulament și care le sînt aplicabile, conform scopului pentru care au fost proiectate respectivele dispozitive.
Secțiunea a 2-a. Libera circulație.
Referirea la standardele în domeniul dispozitivelor
medicale pentru diagnostic in vitro
9. Se admite introducerea pe piață sau punerea în funcțiune a dispozitivelor care sînt conforme cu prevederile prezentului Regulament și a celor care poartă marcajul CE sau marcajul de conformitate SM prevăzut de prezentul Regulament, marcaj ce semnifică faptul că aceste dispozitive au fost supuse evaluării conformității potrivit prevederilor capitolului IV din prezentul Regulament.Referirea la standardele în domeniul dispozitivelor
medicale pentru diagnostic in vitro
10. Pentru dispozitivele destinate evaluării performanței pentru laboratoarele și instituțiile indicate în declarația prevăzută în anexa nr. 8 la prezentul Regulament nu sînt restricții de introducere pe teritoriul Republicii Moldova dacă satisfac condițiile prevăzute în prezentul Regulament.
11. Prevederile prezentului Regulament nu se aplică dispozitivelor destinate expunerii în cadrul tîrgurilor, expozițiilor, demonstrațiilor, întrunirilor științifice și tehnice organizate pe teritoriul Republicii Moldova, cu condiția ca acestea să nu fie utilizate pentru efectuarea analizelor pe probe prelevate de la participanți și să existe o indicație vizibilă că aceste dispozitive nu pot fi comercializate sau puse în funcțiune înainte de a deveni conforme cu prezentul Regulament.
12. Pentru asigurarea unei utilizări sigure și corecte a dispozitivului, informațiile prevăzute în anexa nr. 1 partea В, secțiunea a 9-a se furnizează în limba de stat a Republicii Moldova.
13. La aplicarea prevederilor pct. 12 din prezentul Regulament se ține cont de principiul proporționalității, în mod special:
1) de faptul dacă informațiile trebuie să fie furnizate prin simboluri armonizate, coduri sau alte măsuri notificate;
2) de tipul de utilizator anticipat pentru respectivul dispozitiv.
14. Înregistrările și corespondența privind aplicarea procedurilor de evaluare a conformității se prezintă în limba de stat sau într-o limbă acceptată de organismul notificat de evaluare a conformității.
15. În cazul în care dispozitivele intră sub incidența altor reglementări care, de asemenea, prevăd aplicarea marcajului de conformitate, acesta trebuie să indice conformitatea dispozitivelor cu prevederile tuturor reglementărilor aplicabile.
16. Dacă una sau mai multe reglementări menționate la pct. 15 din prezentul Regulament permit producătorului, pentru o perioadă tranzitorie, să aleagă reglementările pe care să le aplice, marcajul CE semnifică faptul că dispozitivele satisfac numai prevederile acelor reglementări tehnice care sînt aplicate de producător.
17. În acest caz, elementele de identificare particulare ale reglementărilor tehnice aplicate trebuie să fie indicate în documentele, notele sau instrucțiunile care însoțesc dispozitivul.
18. Se consideră că cerințele esențiale prevăzute în anexa nr. 1 la prezentul Regulament sînt îndeplinite dacă dispozitivele medicale sînt conforme cu specificațiile tehnice din standardele europene armonizate, adoptate în calitate de standarde moldovenești din domeniul dispozitivelor medicale (în continuare – standarde).
19. Lista standardelor din domeniul dispozitivelor medicale pentru diagnostic in vitro se aprobă prin ordin de către Agenție și se publică în Monitorul Oficial al Republicii Moldova. Această listă se actualizează ori de cîte ori este cazul.
20. În situația în care Agenția constată că standardele nu respectă în întregime cerințele esențiale prevăzute în anexa nr. 1 la prezentul Regulament, aceasta aplică prevederile prezentului Regulament.
21. Se consideră conforme cu cerințele esențiale stipulate în anexa nr. 1 la prezentul Regulament dispozitivele medicale pentru diagnostic in vitro proiectate și fabricate în conformitate cu specificațiile tehnice generale elaborate pentru dispozitivele incluse în listele A și B din anexa nr. 2 la prezentul Regulament. Specificațiile tehnice generale sînt aprobate prin ordin al ministrului sănătății, muncii și protecției sociale și stabilesc criteriile de evaluare și de reevaluare a performanței, criteriile de eliberare a loturilor, metodele și materialele de referință.
22. Producătorii trebuie să se conformeze specificațiilor tehnice generale sau, după caz, să asigure condiții echivalente specificațiilor tehnice generale în vigoare. Standardele cu referire la dispozitivele medicale pentru diagnostic in vitro includ și specificațiile tehnice generale.
Capitolul III
CLAUZA DE SIGURANȚĂ
23. În situația în care se constată că dispozitivele prevăzute la pct. 9 din prezentul Regulament, corect instalate, întreținute și utilizate conform scopului propus, pot compromite sănătatea și/sau securitatea pacienților, a utilizatorilor și/sau, după caz, a altor persoane ori siguranța proprietății, Agenția aplică măsurile necesare în conformitate cu prevederile Legii nr. 102 din 9 iunie 2017 cu privire la dispozitivele medicale. Producătorul este responsabil de activitățile ulterioare retragerii/interzicerii dispozitivelor medicale.CLAUZA DE SIGURANȚĂ
24. Agenția informează părțile interesate de măsurile întreprinse în conformitate cu prevederile Legii nr. 102 din 9 iunie 2017 cu privire la dispozitivele medicale, invocînd motive întemeiate, în mod special dacă neconformitatea cu prezentul Regulament se datorează:
1) neîndeplinirii cerințelor esențiale prevăzute în anexa nr. 1 la prezentul Regulament;
2) aplicării incorecte a standardelor prevăzute la pct. 18 din prezentul Regulament, în măsura în care se pretinde că standardele au fost aplicate;
3) unor deficiențe ale standardelor.
25. Dacă un dispozitiv neconform poartă marcajul CE, Agenția ia măsurile corespunzătoare în conformitate cu prevederile Legii nr. 102 din 9 iunie 2017 cu privire la dispozitivele medicale.
Capitolul IV
PROCEDURI DE EVALUARE A CONFORMITĂȚII
26. În cazul tuturor dispozitivelor, cu excepția celor cuprinse în anexa nr. 2 la prezentul Regulament și a dispozitivelor pentru evaluarea performanței, în scopul aplicării marcajului de conformitate, producătorul aplică procedura menționată în anexa nr. 3 la prezentul Regulament și emite declarația de conformitate înainte de introducerea pe piață a dispozitivelor. Producătorul trebuie să îndeplinească cerințele suplimentare stabilite în pct. 6 din anexa nr. 3 la prezentul Regulament. În locul acestei proceduri, producătorul poate aplica procedura prevăzută la pct. 28 sau 29.PROCEDURI DE EVALUARE A CONFORMITĂȚII
27. În cazul dispozitivelor medicale pentru autotestare, înainte de a emite declarația de conformitate menționată la pct. 26 din prezentul Regulament, producătorul trebuie să îndeplinească cerințele suplimentare stabilite în pct. 6 din anexa nr. 3 la prezentul Regulament. În locul acestei proceduri, producătorul poate aplica procedura prevăzută la pct. 29 sau 30 din prezentul Regulament.
28. În cazul tuturor dispozitivelor cuprinse în lista A din anexa nr. 2 la prezentul Regulament, cu excepția celor destinate evaluării performanței, producătorul, în scopul aplicării marcajului CE, recurge la una dintre următoarele proceduri referitoare la:
1) declarația de conformitate CE stabilită în anexa nr. 4 la prezentul Regulament;
2) examinarea CE de tip stabilită în anexa nr. 5, asociată cu procedura referitoare la declarația de conformitate, conform anexei nr. 7 la prezentul Regulament privind asigurarea calității producției.
29. În cazul tuturor dispozitivelor cuprinse în lista B din anexa nr. 2 la prezentul Regulament, cu excepția celor destinate evaluării performanței, producătorul, în scopul aplicării marcajului de conformitate, apelează la una dintre următoarele proceduri referitoare la:
1) declarația de conformitate CE stabilită în anexa nr. 4 la prezentul Regulament;
2) examinarea CE de tip stabilită în anexa nr. 5 la prezentul Regulament, asociată cu:
a) procedura privind verificarea CE stabilită în anexa nr. 6 la prezentul Regulament;
b) procedura referitoare la declarația de conformitate CE menționată în anexa nr. 7 la prezentul Regulament.
30. În cazul dispozitivelor de evaluare a performanței, producătorul aplică procedura stabilită în anexa nr. 8 la prezentul Regulament și emite declarația prevăzută în această anexă înainte ca dispozitivele respective să devină disponibile.
Dispozițiile prezentului punct nu afectează reglementările naționale referitoare la aspectele etice ale studiilor de evaluare a performanței dispozitivelor pentru a căror fabricare sînt folosite țesuturi sau substanțe de origine umană.
31. În timpul procedurii de evaluare a conformității unui dispozitiv, producătorul și, dacă este implicat, organismul de certificare notificat trebuie să țină cont de rezultatele obținute în urma oricăror operațiuni de evaluare și verificare efectuate, după caz, în concordanță cu prezentul Regulament, într-o fază intermediară de fabricație.
32. Producătorul poate da instrucțiuni reprezentantului său autorizat în sensul începerii procedurilor prevăzute în anexele nr. 3, nr. 5, nr. 6 și nr. 8 din prezentul Regulament.
33. Producătorul trebuie să păstreze timp de 5 ani de la data fabricării ultimului produs declarația de conformitate, documentația tehnică menționată în anexele nr. 3-8 la prezentul Regulament, precum și hotărîrile, rapoartele și certificatele emise de organismele notificate și, în caz de necesitate, să le pună la dispoziția Agenției pentru inspectare.
Dacă producătorul nu este stabilit în Republica Moldova, obligația de a pune la dispoziție, la cerere, documentația menționată la primul alineat al prezentului punct îi revine reprezentantului său autorizat.
34. Atunci cînd procedura de evaluare a conformității implică intervenția unui organism de certificare notificat, producătorul ori reprezentantul său autorizat se adresează unui organism la alegere, corespunzător sarcinilor în legătură cu care acesta a fost notificat.
35. Organismul notificat cere, în cazurile întemeiate, orice informații sau date necesare pentru stabilirea și menținerea atestării conformității în funcție de procedura aleasă.
36. Deciziile adoptate de organismul notificat în conformitate cu anexele nr. 3-nr.5 la prezentul Regulament au valabilitatea stabilită în Legea nr. 235 din 1 decembrie 2011 privind activitățile de acreditare și de evaluare a conformității.
37. Înregistrările și corespondența referitoare la procedurile prevăzute la pct. 26-29 din prezentul Regulament se redactează în limba de stat.
38. Agenția poate permite, în baza unor informații justificate, înregistrarea în Registrul de stat al dispozitivelor medicale, în scopul introducerii pe piață și al punerii în funcțiune în Republica Moldova, a dispozitivelor medicale pentru care nu s-au efectuat procedurile de evaluare a conformității și a căror utilizare este în interesul protecției sănătății.
39. Prevederile prezentului capitol se aplică în mod corespunzător oricărei persoane fizice sau juridice care produce dispozitivele ce fac obiectul prezentului Regulament și care, fără să le introducă pe piață, le pune în funcțiune și le utilizează în cadrul activității sale profesionale.
Capitolul V
PROCEDURA DE ÎNREGISTRARE
A PRODUCĂTORILOR ȘI DISPOZITIVELOR
ȘI PROCEDURA DE VIGILENȚĂ
Secțiunea 1. Procedura de înregistrare a producătorilor și a dispozitivelor
40. Producătorii care au sediul în Republica Moldova și introduc pe piață dispozitivele sub propriul lor nume au obligația să le înregistreze la Agenție și să comunice următoarele:PROCEDURA DE ÎNREGISTRARE
A PRODUCĂTORILOR ȘI DISPOZITIVELOR
ȘI PROCEDURA DE VIGILENȚĂ
Secțiunea 1. Procedura de înregistrare a producătorilor și a dispozitivelor
1) adresa sediului juridic/domiciliul;
2) informațiile despre reactivi, produși de reacție și materiale de calibrare și control, în ceea ce privește caracteristicile tehnologice generale și/sau substanțele de analizat și orice modificare semnificativă a acestora, inclusiv suspendarea introducerii pe piață;
3) în cazul dispozitivelor care fac obiectul anexei nr. 2 la prezentul Regulament și al dispozitivelor de autotestare, parametrii analitici și, după caz, parametrii de diagnostic conform anexei nr. 1 partea A pct. 3, rezultatele evaluării performanței conform anexei nr. 8 la prezentul Regulament, datele despre certificate și orice modificări semnificative ale acestora, inclusiv sistarea introducerii pe piață a dispozitivelor.
Secțiunea a 2-a. Procedura de vigilență. Baza de date
41. Agenția ia măsurile necesare, în conformitate cu prevederile Legii nr. 102 din 9 iunie 2017 cu privire la dispozitivele medicale, pentru a asigura înregistrarea și evaluarea oricărei informații integre aduse la cunoștință în legătură cu următoarele incidente în care au fost implicate dispozitivele cu marcaj de conformitate:1) orice disfuncție sau orice alterare/depreciere a caracteristicilor și/sau a performanțelor unui dispozitiv medical pentru diagnostic in vitro, precum și orice etichetare, prospect și instrucțiune inadecvate, susceptibile să producă sau că au produs, direct ori indirect, decesul sau afectarea severă a stării de sănătate a unui pacient ori utilizator;
2) orice argument de ordin tehnic sau medical în legătură cu caracteristicile ori performanțele unui dispozitiv la care se referă subpct. 1) din prezentul punct și care ar conduce la o retragere sistematică de pe piață de către producător a dispozitivelor de același tip.
42. Obligația de a anunța Agenția despre incidentele în utilizarea dispozitivelor specificate în prezentul Regulament îi revine producătorilor, reprezentanților autorizați, utilizatorilor, persoanelor juridice ce comercializează dispozitive medicale și importatorilor în conformitate cu prevederile Legii nr. 102 din 9 iunie 2017 cu privire la dispozitivele medicale.
43. După efectuarea unei evaluări, dacă este posibil, împreună cu producătorul și fără a prejudicia aplicarea prevederilor pct. 23 din prezentul Regulament, Agenția informează părțile interesate cu privire la incidentele specificate în prezentul Regulament, pentru care s-au luat sau trebuie să se ia măsuri corespunzătoare, în conformitate cu prevederile Legii nr. 102 din 9 iunie 2017 cu privire la dispozitivele medicale, inclusiv retragerea.
44. Agenția informează, la cerere, autoritățile competente din alte state, cu care are încheiate acorduri cu privire la aspectele menționate la pct. 41-43 din prezentul Regulament și stabilește procedurile de aplicare a prevederilor din prezenta secțiune.
45. Datele înregistrate sînt stocate în baza de date, astfel încît să permită Agenției îndeplinirea atribuțiilor sale conform prezentului Regulament.
Normele de procedură pentru aplicarea prevederilor prezentului punct se aprobă prin ordin al ministrului sănătății, muncii și protecției sociale.
46. Baza de date va cuprinde:
1) datele referitoare la înregistrarea producătorilor și a dispozitivelor, potrivit secțiunii 1 a prezentului capitol;
2) datele referitoare la certificatele emise, modificate, suplimentate, suspendate, retrase sau respinse conform procedurilor prevăzute în anexele nr. 3-nr. 7 la prezentul Regulament;
3) datele obținute potrivit procedurii de vigilență prevăzute în prezenta secțiune la prezentul capitol.
Datele menționate se furnizează în format standard.
47. Procedurile pentru punerea în aplicare a prezentului capitol se adoptă în conformitate cu procedura de reglementare prevăzută de prezentul Regulament.
Capitolul VI
MĂSURI DE MONITORIZARE A SĂNĂTĂȚII PUBLICE
48. Agenția poate adopta toate măsurile temporare necesare și justificate în conformitate cu prevederile Legii nr. 102 din 9 iunie 2017 cu privire la dispozitivele medicale, restrîngerea sau interzicerea unui anumit dispozitiv sau grup de dispozitive, în cazul în care acesta nu întrunește prevederile prezentului Regulament și poate compromite protecția sănătății și/sau sănătatea publică.MĂSURI DE MONITORIZARE A SĂNĂTĂȚII PUBLICE
49. Agenția informează părțile interesate despre măsurile aplicate conform prevederilor din prezentul Regulament.
50. Măsurile necesare pentru modificarea elementelor neesențiale ale prezentului Regulament se adoptă în conformitate cu procedura de reglementare prevăzută de prezentul Regulament.
Capitolul VII
ORGANISME DE EVALUARE A CONFORMITĂȚII NOTIFICATE
51. Ministerul Sănătății, Muncii și Protecției Sociale recunoaște organismele acreditate în conformitate cu prevederile din Legea nr. 235 din 1 decembrie 2011 privind activitățile de acreditare și de evaluare a conformității, pentru realizarea procedurilor de evaluare a conformității a dispozitivelor medicale pentru diagnostic in vitro.ORGANISME DE EVALUARE A CONFORMITĂȚII NOTIFICATE
52. Ministerul Sănătății, Muncii și Protecției Sociale poate retrage recunoașterea unui organism în cazul în care constată că acest organism nu mai respectă criteriile menționate în anexa nr. 9 la prezentul Regulament.
53. Evaluarea conformității dispozitivelor medicale se efectuează de organisme de evaluare a conformității acreditate în condițiile Legii nr. 235 din 1 decembrie 2011 privind activitățile de acreditare și de evaluare a conformității și recunoscute de Ministerul Sănătății, Muncii și Protecției Sociale conform criteriilor stabilite de cadrul normativ în vigoare.
54. Organismul notificat și producătorul ori reprezentantul său autorizat stabilesc, de comun acord, termenele-limită pentru finalizarea activităților de evaluare și verificare prevăzute în anexele nr. 3-7 la prezentul Regulament.
55. Organismul notificat informează celelalte organisme și Ministerul Sănătății, Muncii și Protecției Sociale despre toate certificatele suspendate sau retrase și, la cerere, despre certificatele emise ori respinse. La cererea Ministerului Sănătății, Muncii și Protecției Sociale, pune la dispoziția lor toată informația suplimentară relevantă.
56. În cazul în care se constată că cerințele din prezentul Regulament nu au fost îndeplinite sau au încetat să mai fie îndeplinite de către producător ori dacă un certificat nu ar fi trebuit să fie emis, organismul notificat, ținînd cont de principiul proporționalității, suspendă sau retrage certificatul emis ori impune restricții asupra producătorului pînă în momentul în care conformitatea cu aceste cerințe este asigurată.
57. În cazul suspendării sau retragerii certificatului ori al impunerii de restricții asupra producătorului ori în cazurile în care este necesară o intervenție din partea Ministerului Sănătății, Muncii și Protecției Sociale, organismul notificat informează Ministerul Sănătății, Muncii și Protecției Sociale cu privire la acest fapt.
58. Organismul notificat furnizează, la cerere, toată informația și documentele relevante, pentru a da posibilitate Ministerului Sănătății, Muncii și Protecției Sociale să verifice îndeplinirea cerințelor prevăzute în anexa nr. 9 la prezentul Regulament.
Capitolul VIII
MARCAJUL CE
Secțiunea 1. Marcaj CE
59. Dispozitivele considerate că satisfac cerințele esențiale prevăzute la pct. 8 din prezentul Regulament, cu excepția celor destinate evaluării performanței, trebuie să poarte marcajul CE în momentul introducerii lor pe piață.MARCAJUL CE
Secțiunea 1. Marcaj CE
60. Marcajul CE, conform anexei nr. 10 la prezentul Regulament, trebuie să fie aplicat într-o formă vizibilă, clară și care nu se poate șterge, atît pe dispozitiv sau pe ambalajul său steril, unde este posibil, cît și pe instrucțiunile de utilizare:
1) marcajul CE trebuie să fie aplicat și pe ambalajul în care se comercializează dispozitivul;
2) marcajul CE este însoțit de numărul de identificare al organismului notificat care poartă răspunderea pentru aplicarea procedurilor prevăzute în anexele nr. 3, nr. 4, nr. 6 și nr. 7 la prezentul Regulament.
61. Este interzisă aplicarea de simboluri sau de inscripții care pot induce în eroare terțe părți cu privire la înțelesul ori forma grafică a marcajului CE. Se admite aplicarea oricărui alt semn pe dispozitiv, pe ambalajul său ori pe instrucțiunile care însoțesc dispozitivul, cu condiția ca acesta să nu afecteze vizibilitatea și claritatea marcajului CE.
Secțiunea a 2-a. Marcaj CE aplicat incorect
62. În cazul în care Agenția stabilește că marcajul CE a fost aplicat în mod greșit, producătorul ori reprezentantul său autorizat este obligat să pună capăt acestei situații de încălcare a normelor în domeniu, în condițiile impuse.63. Dacă se menține situația de neconformitate prevăzută la pct. 62 din prezentul Regulament, Agenția adoptă toate măsurile necesare, în conformitate cu prevederile Legii nr. 102 din 9 iunie 2017 cu privire la dispozitivele medicale, pentru restrîngerea sau interzicerea introducerii pe piață a dispozitivului în cauză și/sau, după caz, pentru retragerea lui de pe piață.
64. Dispozițiile pct. 62 și 63 din prezentul Regulament se aplică și în cazul în care marcajul CE s-a aplicat inadecvat pe produse care nu fac obiectul prezentului Regulament.
Capitolul IX
OBLIGAȚIA DE CONFIDENȚIALITATE
65. Persoanele juridice și fizice implicate în îndeplinirea prezentului Regulament sînt obligate să asigure confidențialitatea informațiilor obținute în procesul exercitării sarcinilor lor de serviciu, cu respectarea legislației în vigoare și a practicii naționale cu privire la secretul actului medical și comercial.OBLIGAȚIA DE CONFIDENȚIALITATE
66. Prevederile pct. 65 din prezentul Regulament nu se referă la obligațiile ce le revin Agenției și organismelor notificate cu privire la informarea reciprocă și difuzarea avertismentelor sau obligațiilor persoanelor implicate în furnizarea informațiilor sub incidența legislației penale în vigoare.
Anexa nr. 1
la Regulamentul privind condițiile de introducere
pe piață a dispozitivelor medicale
pentru diagnostic in vitro
la Regulamentul privind condițiile de introducere
pe piață a dispozitivelor medicale
pentru diagnostic in vitro
CERINȚE ESENȚIALE
A. Cerințe generale
1. Dispozitivele trebuie să fie proiectate și fabricate astfel încît, fiind utilizate în condițiile și pentru scopul propus, să nu compromită, direct sau indirect, starea clinică ori siguranța pacienților, siguranța și sănătatea celor care le utilizează sau, după caz, a altor persoane ori siguranța proprietății. Riscul asociat folosirii dispozitivelor trebuie să rămînă în limitele acceptabile în raport cu beneficiul adus pacientului și să aibă un nivel ridicat de siguranță și de protecție a sănătății.A. Cerințe generale
2. Soluțiile adoptate de către producător pentru proiectarea și fabricarea dispozitivelor trebuie să fie conforme cu principiile de siguranță, ținîndu-se cont de nivelul actual de dezvoltare a tehnologiei în domeniu. Pentru selectarea celor mai potrivite soluții, producătorul va aplica următoarele principii:
1) eliminarea sau reducerea la maximum a riscurilor prin proiectare și realizare sigură;
2) asigurarea măsurilor de protecție, acolo unde este cazul, în legătură cu riscurile care nu pot fi eliminate;
3) informarea utilizatorilor despre riscurile persistente, datorate insuficientelor măsuri de protecție adoptate.
3. Dispozitivele trebuie să fie proiectate și realizate în așa fel încît să fie adecvate scopurilor specificate de către producător, avînd în vedere nivelul actual de dezvoltare a tehnologiei în domeniu. Dispozitivele trebuie să realizeze următoarele performanțe: sensibilitate analitică, sensibilitate de diagnostic, specificitate analitică, specificitate de diagnostic, acuratețe, repetabilitate, reproductibilitate, inclusiv controlul interferențelor cunoscute și al limitelor de detecție stabilite de către producător. Trasabilitatea valorilor atribuite etaloanelor și/sau materialelor de control se asigură prin proceduri de măsurare de referință și/sau materiale de referință disponibile de calitate superioară.
4. Caracteristicile și performanțele specificate la pct. 1 și 3 din prezenta anexă nu trebuie să fie afectate în asemenea măsură încît să compromită sănătatea sau siguranța pacientului, a utilizatorului și, după caz, a altor persoane, pe întreaga durată de funcționare indicată de producător, atunci cînd dispozitivul este supus unor suprasolicitări ce pot interveni în timpul funcționării în condiții normale de exploatare. Dacă nu se specifică durata de funcționare, prevederea se aplică pentru durata de utilizare apreciată pentru un dispozitiv de acel tip, avînd în vedere scopul propus și utilizarea prevăzută a dispozitivului.
5. Dispozitivele trebuie să fie proiectate, fabricate și ambalate astfel încît caracteristicile și performanțele lor în timpul folosirii să nu fie afectate ca urmare a condițiilor de transport și de depozitare (temperatură, umiditate etc.), ținînd cont de instrucțiunile și informațiile furnizate de producător.
B. Cerințe cu privire la proiectare și construcție
Secțiunea 1. Proprietăți chimice și fizice
6. Dispozitivele trebuie să fie proiectate și fabricate astfel încît să posede caracteristicile și performanțele cuprinse în partea A din prezenta anexă. Se va urmări în mod special ca performanța analitică să nu fie afectată din cauza incompatibilității materialelor folosite și a probelor (celule, țesuturi, fluide biologice și microorganisme) destinate utilizării împreună cu dispozitivul, conform scopului propus al acestuia.Secțiunea 1. Proprietăți chimice și fizice
7. Dispozitivele trebuie să fie proiectate, fabricate și ambalate astfel încît să minimizeze riscul produs de scurgeri și contaminări cu reziduuri pentru persoanele implicate în transportul, depozitarea și utilizarea acestora, conform scopului propus.
Secțiunea a 2-a. Infecția și contaminarea microbiană
8. Dispozitivele și procesul de fabricație trebuie să fie astfel concepute încît să elimine sau să reducă la minimum riscul de infecție a utilizatorului și a terțelor persoane. Proiectarea trebuie să permită manipularea ușoară și să micșoreze contaminarea dispozitivului sau scurgerile din acesta în timpul folosirii, iar în cazul recipientelor pentru probe, riscul contaminării probei. Procesul de fabricație trebuie să fie adecvat acestor scopuri.9. Dacă un dispozitiv încorporează o substanță biologică, riscul de infecție trebuie să fie redus la minimum prin selectarea de donatori adecvați și de substanțe corespunzătoare, precum și prin utilizarea de proceduri adecvate validate de inactivare, conservare, testare și control.
10. Dispozitivele etichetate ca „STERILE” sau într-o stare microbiologică specială trebuie să fie proiectate, fabricate și ambalate pentru a se asigura păstrarea lor în starea microbiologică indicată pe etichetă atunci cînd sînt introduse pe piață, în condițiile de transport și depozitare specificate de către producător, dacă ambalajul nu este deschis sau deteriorat.
11. Dispozitivele etichetate ca „STERILE” sau într-o stare microbiologică specială trebuie să fie fabricate printr-o metodă corespunzătoare validată.
12. Sistemele de ambalare pentru dispozitive, altele decît cele menționate la pct. 10 din prezenta anexă, trebuie să păstreze produsul fără a-l deteriora, la nivelul de curățenie indicat de producător și, dacă dispozitivele trebuie să fie sterilizate înainte de utilizare, să reducă la minimum riscul de contaminare microbiană.
Este necesar să se ia măsuri pentru a se reduce la minimum riscul de contaminare microbiană în cursul selecției și al mînuirii de materii prime, al fabricării, al depozitării și al distribuției, dacă performanța dispozitivului este afectată de o astfel de contaminare.
13. Dispozitivele destinate sterilizării trebuie să fie fabricate în condiții (de exemplu, de mediu) corespunzătoare.
14. Sistemele de ambalare pentru dispozitivele nesterile trebuie să protejeze produsul de deteriorări, păstrîndu-se nivelul de curățenie prevăzut. La dispozitivele ce trebuie să fie sterilizate anterior folosirii trebuie redus riscul de contaminare microbiană; sistemul de ambalare trebuie să fie adecvat, conform metodei de sterilizare indicate de producător.
Secțiunea a 3-a. Proprietățile de construcție și mediul
15. Dacă dispozitivele sînt destinate folosirii în combinație cu alte dispozitive sau echipamente, întreaga combinație, inclusiv sistemul de conectare, trebuie să fie sigură și să nu afecteze performanțele specificate ale dispozitivelor. Orice restricții de utilizare trebuie să fie indicate pe etichetă sau în instrucțiunile de folosire.16. Dispozitivele trebuie să fie proiectate și fabricate astfel încît să reducă la minimum riscurile legate de utilizarea lor împreună cu materiale, substanțe și gaze cu care ar putea veni în contact în condiții normale de utilizare.
17. Dispozitivele trebuie să fie proiectate și fabricate astfel încît să elimine sau să reducă cît mai mult posibil:
1) riscul de lezare legat de caracteristicile fizice, inclusiv creșterile de volum/presiune/dimensiuni și caracteristicile ergonomice;
2) riscurile legate de influențe externe previzibile, cum ar fi: cîmpurile magnetice, efectele electrice externe, descărcările electrostatice, temperatura, umiditatea, presiunea și variațiile de presiune sau de accelerație ori pătrunderile accidentale de substanțe în dispozitiv.
Dispozitivele trebuie să fie proiectate și fabricate astfel încît să ofere un nivel adecvat de imunitate intrinsecă la perturbațiile electromagnetice, pentru a putea acționa conform scopului propus.
18. Dispozitivele trebuie să fie proiectate și fabricate în așa fel încît să reducă riscurile de incendiu sau explozie în timpul folosirii normale și în condiții de defectare simplă. O atenție deosebită trebuie să fie acordată dispozitivelor a căror utilizare proiectată presupune expunerea la sau folosirea lor în asociație cu substanțe inflamabile ori substanțe care întrețin arderea.
19. Dispozitivele trebuie să fie proiectate și fabricate astfel încît să faciliteze distrugerea în siguranță a reziduurilor.
20. Scara de măsurare, monitorizare sau afișare (inclusiv modificarea culorilor și a altor indicatori vizuali) trebuie să fie proiectată și fabricată în conformitate cu principiile ergonomice, conform scopului propus al dispozitivului.
Secțiunea a 4-a. Dispozitive cu funcție de măsurare
21. Dispozitivele cu funcție de măsurare analitică trebuie să fie proiectate și fabricate astfel încît să asigure o stabilitate și precizie corespunzătoare măsurilor, în limitele specificate, ținîndu-se cont de scopul propus al dispozitivului și de procedurile și materialele de referință disponibile. Limitele de precizie trebuie să fie indicate de producător.22. Determinările realizate de dispozitivul cu funcție de măsurare vor fi exprimate în unități de măsură legale.
Secțiunea a 5-a. Protecția împotriva iradierii
23. Dispozitivele trebuie să fie proiectate, fabricate și ambalate astfel încît expunerea utilizatorilor și a altor persoane la radiația emisă să fie minimizată.24. Dacă dispozitivele sînt destinate să emită radiații potențial periculoase, vizibile și/sau invizibile, ele necesită să fie pe cît posibil:
1) proiectate și realizate astfel încît să asigure că parametrii și cantitatea radiației emise pot fi controlate și/sau ajustate;
2) prevăzute cu afișaj vizual și/sau avertizare sonoră a acestor emisii.
25. Instrucțiunile de operare pentru dispozitivele ce emit radiații trebuie să ofere informații asupra naturii radiației emise, a mijloacelor de protecție a utilizatorului și a măsurilor ce pot fi întreprinse pentru evitarea utilizării necorespunzătoare și pentru eliminarea riscului inerent la instalare.
Secțiunea a 6-a. Cerințe pentru dispozitive medicale
conectate la o sursă de energie sau echipate cu o sursă de energie
26. Dispozitivele care încorporează sisteme electronice programabile, inclusiv software, trebuie să fie proiectate astfel încît să asigure repetabilitatea, siguranța și performanța acestor sisteme în raport cu scopul propus.conectate la o sursă de energie sau echipate cu o sursă de energie
27. Dispozitivele trebuie să fie proiectate și realizate astfel încît să reducă pe cît posibil riscurile de generare a perturbațiilor electromagnetice, care ar putea afecta funcționarea altor dispozitive sau echipamente din mediul înconjurător uzual.
28. Dispozitivele trebuie să fie proiectate și fabricate astfel încît să evite pe cît posibil riscul șocului electric accidental în timpul folosirii normale și în condiții de prim defect, atunci cînd dispozitivele sînt instalate și întreținute corect.
Secțiunea a 7-a. Protecția împotriva riscurilor mecanice și termice
29. Dispozitivele trebuie să fie proiectate și fabricate astfel încît să protejeze utilizatorul împotriva riscurilor mecanice. Dispozitivele trebuie să fie suficient de stabile în condiții de funcționare prevăzute. Ele trebuie să fie rezistente la solicitările inerente în mediul de lucru prevăzut și să-și păstreze rezistența pe parcursul perioadei de funcționare proiectate, cu condiția respectării cerințelor de întreținere și inspecție indicate de producător. Dacă există riscuri determinate de prezența pieselor în mișcare, rupere sau detașare ori pierderi de substanțe, trebuie să fie incluse mijloace adecvate de protecție. Mijloacele de protecție sau cele atașate dispozitivului pentru asigurarea protecției, în special împotriva pieselor în mișcare, trebuie să fie sigure și să nu împiedice accesul pentru funcționarea normală a dispozitivului sau să împiedice întreținerea de rutină a dispozitivului așa cum a fost prevăzută de producător.30. Dispozitivele trebuie să fie proiectate și fabricate astfel încît să reducă la minimum riscurile ce apar din vibrația generată de dispozitive, bazîndu-se pe progresele tehnice și pe mijloacele disponibile pentru reducerea vibrației, în special la sursa de alimentare, numai dacă obținerea vibrațiilor nu reprezintă scopul propus.
31. Dispozitivele trebuie să fie proiectate și fabricate astfel încît să reducă cît se poate de mult riscurile care apar din zgomotul emis, în special la sursa de alimentare, dacă acest zgomot nu este parte a scopului propus.
32. Terminalele și conectorii alimentați de la o sursă de electricitate, de gaz, hidraulică sau pneumatică și care trebuie să fie mînuiți de utilizator trebuie să fie proiectați astfel încît să minimizeze toate riscurile posibile.
33. Părțile accesibile ale dispozitivelor (cu excepția părților sau a suprafețelor proiectate pentru a emite căldură sau pentru a atinge temperaturi prestabilite) și împrejurimile acestora nu trebuie să atingă temperaturile potențial periculoase în condiții de utilizare normală.
Secțiunea a 8-a. Cerințe pentru dispozitivele destinate autotestării
34. Dispozitivele pentru autotestare trebuie să fie proiectate și fabricate conform condițiilor de îndeplinire a scopului prevăzut, ținînd seama de îndemînarea și de mijloacele disponibile ale celor care le utilizează, precum și de influența care rezultă din variațiile ce pot fi rațional anticipate în priceperea utilizatorilor și condițiile de mediu. Instrucțiunile și informațiile furnizate de producător trebuie să se înțeleagă și să se aplice ușor de către utilizator.35. Dispozitivele pentru autotestare trebuie să fie proiectate și realizate astfel încît:
1) să asigure utilizarea ușoară a dispozitivului de către utilizator pe parcursul tuturor etapelor procedurii;
2) să reducă pe cît posibil riscul erorilor de manevrare a dispozitivului și de interpretare a rezultatelor.
36. Dispozitivele pentru autotestare urmează să prevadă controlul utilizatorului, cum ar fi procedura prin care utilizatorul verifică în timpul utilizării dacă produsul îndeplinește scopul prevăzut.
Secțiunea a 9-a. Informații furnizate de producător
37. Fiecare dispozitiv trebuie să fie însoțit de informațiile necesare pentru a identifica producătorul și pentru a fi folosit în siguranță, ținînd seama de pregătirea și de cunoștințele potențialilor utilizatori.Aceste informații trebuie să fie pe etichetă și în instrucțiunile de utilizare.
În măsura în care este posibil și potrivit, informațiile necesare pentru a folosi dispozitivele în siguranță trebuie să fie afișate chiar pe dispozitiv și/sau pe ambalajul în care sînt comercializate. Dacă nu se practică ambalarea individuală, informațiile se redactează într-o broșură furnizată cu unul sau mai multe dispozitive.
Instrucțiunile de folosire trebuie să fie incluse în ambalaj pentru unul sau mai multe dispozitive. În cazuri excepționale, bine justificate, instrucțiunile de folosire nu sînt necesare dacă dispozitivul va fi folosit corect și în siguranță fără aceste instrucțiuni.
38. Dacă este posibil, aceste informații trebuie să ia forma simbolurilor. Orice simbol/culoare de identificare trebuie să fie conform/conformă standardelor. În cazul în care nu există standarde, simbolurile și culorile trebuie să fie descrise în documentația furnizată odată cu dispozitivul.
39. În cazul dispozitivelor care conțin un preparat ce se consideră periculos, ținîndu-se seama de natura și cantitatea constituenților săi și de forma sub care aceștia se prezintă, se aplică cerințele de etichetare și simbolurile de pericol din legislația cu privire la substanțele și preparatele chimice periculoase. Dacă spațiul este insuficient pentru ca toate informațiile să fie plasate pe dispozitiv sau pe etichetă, simbolurile de pericol trebuie să fie plasate pe etichetă, iar celelalte informații se vor conține în instrucțiunile de utilizare.
40. Eticheta trebuie să conțină următoarele date:
1) denumirea și adresa producătorului. Pentru dispozitivele importate în scopul de a fi distribuite în Republica Moldova, eticheta sau ambalajul exterior sau instrucțiunile de folosire vor conține în plus numele și adresa reprezentantului autorizat al producătorului;
2) detaliile strict necesare utilizatorului pentru identificarea fără posibilitate de eroare, a dispozitivului și a conținutului pachetului;
3) cuvîntul „STERIL” sau inscripția cu referire la starea microbiologică specială sau starea de puritate, acolo unde este cazul;
4) numărul lotului precedat de cuvîntul „LOT” sau numărul de serie, după caz;
5) dacă este necesar, data pînă la care dispozitivul poate fi utilizat în siguranță, fără afectarea performanței, exprimată, în ordine, prin anul, luna și, după caz, ziua;
6) în cazul dispozitivelor pentru evaluarea performanței, inscripția „pentru evaluarea performanței”;
7) inscripția că dispozitivul este utilizat in vitro;
8) condițiile speciale de păstrare și/sau de manevrare;
9) instrucțiunile speciale de utilizare;
10) atenționările și/sau precauțiile necesare;
11) dacă dispozitivul este destinat autotestării, precizarea clară a acestui fapt.
41. Dacă scopul propus al dispozitivului nu este evident pentru utilizator, producătorul trebuie să definească, în mod clar, în instrucțiunile de utilizare și, dacă este cazul, pe etichetă, domeniul și funcțiile dispozitivului.
42. Acolo unde este posibil, dispozitivul și părțile sale detașabile trebuie să fie accesibile pentru a fi identificate în numărul de lot și pentru a face posibilă detectarea oricărui risc potențial al dispozitivului și părților sale componente.
43. Instrucțiunile de utilizare trebuie să conțină următoarele:
1) datele prevăzute la pct. 40 din prezenta anexă, cu excepția subpct. 4) și 5);
2) compoziția reactivului, prin natura și cantitatea sau concentrația ingredientelor active ale reactivului (reactivilor) sau kitului (trusei), precum și precizarea, dacă este cazul, că dispozitivul conține alte ingrediente ce ar putea afecta măsurarea;
3) condițiile de depozitare și durata de păstrare după prima deschidere a containerului primar, împreună cu condițiile de depozitare și stabilitate a reactivilor de lucru;
4) performanțele menționate la pct. 3 din prezenta anexă;
5) precizarea oricărui echipament special necesar, incluzînd informații pentru identificarea acelui echipament în vederea utilizării corecte;
6) tipul de probe utilizate, condițiile speciale de recoltare, pretratament și, dacă este necesar, condițiile de depozitare și instrucțiunile pentru pregătirea pacientului;
7) descrierea detaliată a procedurii de urmat la utilizarea dispozitivului;
8) procedura de măsurare care trebuie respectată la utilizarea dispozitivului, inclusiv, dacă este cazul:
a) principiul care stă la baza metodei;
b) caracteristicile de performanță analitică specifice (de exemplu: sensibilitate, specificitate, acuratețe, repetabilitate, reproductibilitate, limite de detectare și intervalul de măsurare, inclusiv informații necesare pentru controlul unor interferențe importante cunoscute), limite ale metodei și informații despre folosirea de către utilizator a unor materiale și proceduri de referință disponibile;
c) detalii asupra oricărei alte proceduri suplimentare sau manipulări necesare înainte de utilizarea dispozitivului (de exemplu: reconstituire, incubare, diluare, verificarea instrumentelor);
d) indicația dacă este necesară o pregătire specială;
9) expresia matematică în baza căreia se calculează rezultatul analitic;
10) măsurările ce trebuie efectuate în cazul modificării performanței analitice a dispozitivului;
11) informațiile necesare utilizatorilor cu privire la:
a) controlul intern al calității, inclusiv procedurile de validare specifice;
b) calibrarea;
12) intervalele de referință pentru cantitățile ce sînt determinate, incluzînd descrierea populației de referință corespunzătoare;
13) dacă dispozitivul trebuie să fie instalat sau conectat la alte dispozitive medicale pentru a funcționa conform scopului propus, detaliile cu privire la caracteristicile sale pentru identificarea dispozitivelor ce necesită să fie utilizate pentru a se obține un sistem corect și sigur;
14) toate informațiile necesare pentru verificarea instalării corespunzătoare a dispozitivului, a utilizării sale corecte și sigure, detaliile asupra naturii și frecvenței operațiunilor de întreținere și calibrare, necesare în vederea asigurării utilizării corecte și sigure pe toată durata de funcționare; informațiile privind distrugerea în siguranță a reziduurilor;
15) detaliile pentru orice tratament sau manevră necesară înainte ca dispozitivul să fie utilizat (de exemplu, sterilizare, asamblare finală);
16) instrucțiunile necesare în caz de deteriorare a ambalajului protector și, dacă este cazul, metoda potrivită de resterilizare sau decontaminare;
17) dacă dispozitivul este reutilizabil, indicațiile privind procedurile necesare pentru reutilizare, inclusiv pentru curățare, dezinfectare, ambalare, resterilizare sau decontaminare, precum și orice restricții privind numărul de reutilizări;
18) precauțiile privind expunerea, în condiții de mediu previzibile, la cîmpuri magnetice, influențe electrice externe, descărcări electrostatice, presiune sau variație de presiune, accelerație și surse de aprindere termică;
19) precauțiile privind orice risc special, neobișnuit legat de utilizarea sau de distrugerea dispozitivului, inclusiv măsuri de protecție speciale. Dacă dispozitivul include substanțe de origine umană sau animală, trebuie avută în vedere natura lor potențial infecțioasă;
20) specificațiile pentru dispozitivele de autotestare:
a) rezultatele trebuie să fie exprimate și prezentate astfel încît să fie înțelese ușor de un neprofesionist; informațiile trebuie să fie însoțite de îndrumări pentru utilizator asupra modului de a acționa (în cazul unui rezultat pozitiv, negativ sau neconcludent) și asupra posibilității unor rezultate fals pozitive sau fals negative;
b) anumite particularități pot fi omise cu condiția ca alte informații furnizate de producător să fie suficiente pentru a permite utilizatorului să folosească dispozitivul și să înțeleagă rezultatele oferite de dispozitiv;
c) informația furnizată trebuie să includă un enunț care să îl determine pe utilizator să nu ia nicio decizie medicală relevantă înainte de a-și consulta medicul curant;
d) informația trebuie să precizeze de asemenea că, în cazul în care dispozitivul destinat autotestării este utilizat pentru monitorizarea unei afecțiuni existente, pacientul trebuie doar să-și adapteze tratamentul, dacă a fost pregătit corespunzător în acest sens;
21) data apariției sau a ultimei revizuiri a instrucțiunilor de utilizare.
Anexa nr. 2
la Regulamentul privind condițiile de introducere
pe piață a dispozitivelor medicale
pentru diagnostic in vitro
la Regulamentul privind condițiile de introducere
pe piață a dispozitivelor medicale
pentru diagnostic in vitro
LISTA
dispozitivelor medicale specificate în punctele 28 și 29
din prezentul Regulament
Lista A
1) Reactivi și produși de reacție, inclusiv calibratori și materiale de referință pentru determinarea următoarelor grupe de sînge: sistem ABO, Rhesus (C, c, D, E, e) anti-Kell;dispozitivelor medicale specificate în punctele 28 și 29
din prezentul Regulament
Lista A
2) reactivi și produși de reacție, inclusiv calibratori și materiale de referință pentru detectarea, confirmarea și cuantificarea în probele prelevate din organismul uman a markerilor infecției cu HIV (HIV 1 și HIV 2), HTLV I și II și virus hepatic B, C și D;
3) teste Varianta bolii Creutzfeldt-Jakob (vCJD) pentru screeningul sîngelui, diagnostic și confirmare.
LISTA B
1) Reactivi și produși de reacție, inclusiv calibratori și materiale de referință pentru determinarea următoarelor grupe de sînge: anti-Duffy și anti-Kidd;2) reactivi și produși de reacție, inclusiv calibratori și materiale de referință pentru determinarea anticorpilor atipici antieritrocitari;
3) reactivi și produși de reacție, inclusiv calibratori și materiale de referință pentru detectarea și cuantificarea în probele prelevate din organismul uman a următoarelor infecții congenitale: rubeola și toxoplasmoza;
4) reactivi și produși de reacție, inclusiv calibratori și materiale de referință pentru diagnosticarea bolii ereditare fenilcetonurie;
5) reactivi și produși de reacție, inclusiv calibratori și materiale de referință pentru determinarea următoarelor infecții la om: citomegalovirus, chlamydia;
6) reactivi și produși de reacție, inclusiv calibratori și materiale de referință pentru determinarea următoarelor grupuri tisulare HLA: DR, A și B;
7) reactivi și produși de reacție, inclusiv calibratori și materiale de referință pentru determinarea markerilor tumorali PSA;
8) reactivi și produși de reacție, inclusiv calibratori, materiale de referință și software, destinate în special evaluării riscului de trisomie 21;
9) dispozitive pentru autodiagnostic, inclusiv, calibratori și materiale de referință pentru măsurarea nivelului de glucide în sînge.
Anexa nr. 3
la Regulamentul privind condițiile de introducere
pe piață a dispozitivelor medicale
pentru diagnostic in vitro
la Regulamentul privind condițiile de introducere
pe piață a dispozitivelor medicale
pentru diagnostic in vitro
DECLARAȚIA DE CONFORMITATE
1. Declarația de conformitate este înscrisul prin care producătorul ori reprezentantul său autorizat care îndeplinește obligațiile impuse la pct. 2-5, și, în cazul produselor destinate autotestării, obligațiile impuse la pct. 6 din prezenta anexă asigură și declară că produsele respective îndeplinesc prevederile aplicabile ale prezentului Regulament. Producătorul trebuie să aplice marcajul în conformitate cu prevederile prezentului Regulament.2. Producătorul trebuie să pregătească documentația tehnică descrisă la pct. 3 din prezenta anexă și să asigure că în procesul de fabricație sînt respectate principiile de asigurare a calității stabilite la pct. 4 din prezenta anexă.
3. Documentația tehnică trebuie să permită evaluarea conformității produsului cu cerințele prezentului Regulament.
Aceasta trebuie să conțină, în special:
1) descrierea generală a produsului, incluzînd toate variantele proiectate;
2) documentația asupra sistemului calității;
3) informația privind proiectarea, inclusiv determinarea caracteristicilor materialelor de bază, a caracteristicilor și limitelor performanței dispozitivelor, metodele de fabricație și, în cazul instrumentelor, schițele de proiect, diagramele componentelor, subansamblurile, circuitele etc.;
4) în cazul dispozitivelor ce conțin țesuturi de origine umană sau substanțe derivate din astfel de țesuturi, informația privind originea acestora și condițiile în care au fost recoltate;
5) descrierile și explicațiile necesare pentru a înțelege caracteristicile, schițele și diagramele menționate mai sus și modul de utilizare a produsului;
6) rezultatele analizei riscurilor și, după caz, lista standardelor menționate în pct. 18 din prezentul Regulament, aplicate în întregime sau în parte, și descrierile soluțiilor adoptate pentru a îndeplini cerințele esențiale ale prezentului Regulament, dacă standardele menționate la pct. 18 nu au fost aplicate în întregime;
7) în cazul produselor sterile sau într-o stare microbiologică de curățenie specială, descrierea procedurilor utilizate;
8) rezultatele calculelor de proiectare și ale inspecțiilor efectuate;
9) dacă dispozitivul urmează să fie asociat cu alte dispozitive pentru a funcționa conform scopului propus, este necesar să fie aduse dovezi că dispozitivul satisface cerințele esențiale atunci cînd este asociat cu oricare dintre aceste dispozitive avînd caracteristicile specificate de producător;
10) rapoartele de încercări;
11) datele adecvate privind evaluarea performanței, care să indice performanțele prevăzute de producător și susținute de un sistem de măsurare de referință (atunci cînd este disponibil), cu informații despre metodele de referință, materialele de referință, valorile de referință cunoscute, precizia și unitățile de măsură utilizate; datele urmează să provină din studiile făcute în condiții clinice sau în alt mediu adecvat ori din referințe biografice relevante;
12) etichetele și instrucțiunile de utilizare;
13) rezultatele studiilor de stabilitate.
4. Producătorul ia măsurile necesare pentru a se asigura că în procesul de fabricație se respectă principiile de asigurare a calității adecvate a produselor fabricate.
Sistemul calității se referă la:
1) structurile organizatorice și responsabilități;
2) procesele de fabricație și controlul sistematic al calității producției;
3) mijloacele de monitorizare a performanțelor sistemului calității.
5. Producătorul instituie și actualizează permanent o procedură sistematică de analiză a experienței obținute în ceea ce privește dispozitivele în faza de postproducție și de aplicare a acțiunilor corective necesare, ținînd cont de natura și de riscurile legate de produs. Acesta trebuie să comunice Agenției următoarele incidente imediat ce a luat cunoștință de ele. La acestea se raportă:
1) orice disfuncție, defectare sau deteriorare a caracteristicilor și/sau a performanțelor dispozitivului, precum și orice neconformitate în etichetare sau în instrucțiunile de utilizare, care, direct ori indirect, ar putea sau ar fi putut conduce la decesul unui pacient, al unui utilizator ori al unei alte persoane sau la deteriorarea severă a stării de sănătate a acestora;
2) orice cauză de ordin tehnic sau medical legată de caracteristicile sau de performanța dispozitivului, care, pentru motivele menționate la subpct. 1) din prezentul punct, conduce la retragerea sistematică a dispozitivelor de același tip de către producător.
6. Pentru dispozitivele destinate autotestării, producătorul trebuie să depună o cerere în scopul examinării proiectului la un organism notificat, ales în conformitate cu prevederile prezentului Regulament.
7. Cererea trebuie să permită înțelegerea proiectului dispozitivului și evaluarea conformității cu cerințele prezentului Regulament aplicabile proiectului.
Ea va include:
1) rapoartele de testare, incluzînd, după caz, rezultatele studiilor realizate cu neprofesioniști;
2) datele ce relevă posibilitatea de manipulare a dispozitivului, ținînd cont de scopul propus pentru autotestare;
3) informațiile ce vor fi furnizate împreună cu dispozitivul pe etichetă și în instrucțiunile de utilizare.
8. Organismul notificat examinează cererea și, dacă proiectul este în conformitate cu prevederile prezentului Regulament, eliberează solicitantului un certificat de examinare a proiectului. Organismul notificat poate cere ca documentația să fie completată cu probe și teste ulterioare, care să permită evaluarea conformității cu cerințele prezentului Regulament aplicabile proiectului. Certificatul conține concluziile examinării, condițiile de validitate, datele necesare pentru identificarea proiectului aprobat și, după caz, descrierea scopului propus al dispozitivului.
9. Solicitantul informează organismul notificat care a eliberat certificatul de examinare a proiectului despre orice modificare semnificativă a proiectului aprobat. Modificările din proiectul aprobat trebuie să fie aprobate ulterior de organismul notificat care a eliberat certificatul de examinare a proiectului, dacă schimbările ar putea afecta conformitatea cu cerințele esențiale ale prezentului Regulament sau cu condițiile prevăzute pentru utilizarea produsului. Această aprobare suplimentară va lua forma unui document adițional la certificatul de examinare a proiectului.
Anexa nr. 4
la Regulamentul privind condițiile de introducere
pe piață a dispozitivelor medicale
pentru diagnostic in vitro
la Regulamentul privind condițiile de introducere
pe piață a dispozitivelor medicale
pentru diagnostic in vitro
DECLARAȚIA DE CONFORMITATE
Sistemul complet de asigurare a calității
1. Producătorul trebuie să asigure aplicarea sistemului de calitate aprobat pentru proiectarea, producerea și inspecția finală a dispozitivelor medicale, așa cum este specificat la pct. 3, și să fie subiectul auditului conform pct. 3 subpct. 3), și al inspecțiilor de supraveghere, conform pct. 5 din prezenta anexă. În plus, producătorul pentru dispozitivele prevăzute în lista A din anexa nr. 2 la prezentul Regulament trebuie să respecte procedurile stabilite la pct. 4 și 6 din prezenta anexă.Sistemul complet de asigurare a calității
2. Declarația de conformitate este procedura prin care producătorul care îndeplinește cerințele impuse la pct. 1 din prezenta anexă asigură și declară că dispozitivele îndeplinesc prevederile prezentului Regulament ce le sînt aplicabile.
Producătorul aplică marcajul în conformitate cu prevederile prezentului Regulament și emite declarația de conformitate referitoare la dispozitivele examinate.
3. Cerințele sistemului calității sînt următoarele:
1) producătorul trebuie să depună la un organism notificat o cerere pentru atestarea sistemului calității, care va include:
a) denumirea sau numele, după caz, și adresa producătorului și ale oricărui loc de fabricație în care se asigură sistemul calității;
b) informațiile relevante cu privire la dispozitiv sau la categoria de dispozitive acoperite de procedură;
c) declarația scrisă în care se menționează că nu a fost depusă o cerere la un alt organism notificat pentru același dispozitiv cu privire la sistemul calității;
d) documentația privind sistemul calității;
e) angajamentul producătorului de a acoperi în totalitate cerințele impuse prin sistemul calității aprobat;
f) angajamentul producătorului de a menține în mod corespunzător sistemul calității aprobat;
g) angajamentul producătorului de a institui și de a ține la zi o procedură sistematică de analiză a experienței obținute cu dispozitivele în faza de postproducție și de a implementa mijloacele adecvate pentru a aplica orice acțiune corectivă necesară sau notificare, după cum se precizează la pct. 5 din anexa nr. 3 la prezentul Regulament.
Sistemul calității trebuie să se refere la:
- structurile organizatorice și responsabilități;
- procesele de fabricație și controlul sistematic al calității producției;
- mijloacele de monitorizare a performanțelor sistemului calității;
2) aplicarea sistemului calității trebuie să asigure că dispozitivele sînt conforme cu prevederile prezentului Regulament ce le sînt aplicabile, la toate etapele, de la proiectare pînă la inspecția finală. Toate elementele, cerințele și prevederile adoptate de producător pentru sistemul său de calitate trebuie să fie documentate în mod sistematic și ordonat sub formă de proceduri și declarații scrise privind politicile de calitate, cum ar fi planuri, programe și înregistrări de calitate.
Acestea trebuie să cuprindă, în special, o descriere adecvată:
a) a obiectivelor producătorului privind calitatea;
b) a modului de organizare a producerii și, în particular:
- structurile organizatorice, responsabilitățile echipei de conducere și a autorității organizatorice în ceea ce privește calitatea proiectării și fabricării dispozitivelor;
- metodele de monitorizare a eficienței sistemului calității și, în particular, capacitatea lui de a atinge calitatea dorită în proiectare și în producere, inclusiv controlul dispozitivelor ce nu se conformează cerințelor de calitate;
c) a procedurilor de monitorizare și de verificare a proiectului dispozitivului și, în special:
- descrierea generală a dispozitivului, inclusiv a tuturor variantelor planificate;
- documentația menționată la pct. 3 subpct. 3)-13) din anexa nr. 3 la prezentul Regulament;
- în cazul dispozitivelor destinate autotestării, informațiile menționate la pct. 6 și 7 din anexa nr. 3 la prezentul Regulament;
- tehnicile utilizate pentru controlul și verificarea proiectului și a proceselor și măsurilor sistematice ce vor fi utilizate în proiectare;
d) a tehnicilor de inspecție și de asigurare a calității în stadiul de producție și, în particular:
- procesele și procedurile care vor fi utilizate, în special cu privire la sterilizare;
- procedurile legate de achiziționare;
- procedurile de identificare a produsului, schițate și actualizate prin proiecte, specificații sau alte elemente relevante, în orice stadiu de producție;
e) a încercărilor și verificărilor adecvate, care trebuie să fie efectuate înainte, în timpul și după fabricație, a frecvenței cu care vor avea loc și a echipamentelor de testare utilizate; trebuie să fie asigurată trasabilitatea măsurărilor.
Producătorul trebuie să realizeze controalele și încercările cerute în conformitate cu nivelul actual de dezvoltare a tehnologiei în domeniu. Controalele și încercările trebuie să acopere procesul fabricației, inclusiv caracteristicile materiilor prime și dispozitivele individuale sau fiecare lot de dispozitive fabricate.
La testarea dispozitivelor prevăzute în lista A din anexa nr. 2 la prezentul Regulament, producătorul urmează să se bazeze pe cele mai recente informații disponibile, în particular în ceea ce privește complexitatea biologică și variabilitatea specimenelor de testat cu dispozitivul pentru diagnostic in vitro respectiv;
3) organismul notificat trebuie să auditeze sistemul calității pentru a stabili conformitatea cu cerințele menționate la subpct. 2) din prezentul punct. Se presupune că sistemul calității care implementează standardele relevante este conform cu aceste cerințe.
Echipa de evaluare trebuie să aibă, în mod obligatoriu, experiență în evaluarea tehnologiilor respective. Procedura de evaluare include inspecția la locul de producție și, în cazuri justificate, la locul de producție al furnizorilor și/sau al subcontractanților, pentru a inspecta procesul de fabricație.
Decizia inspecției, care conține o evaluare argumentată și concluziile inspecției, este comunicată producătorului;
4) producătorul informează organismul notificat care a aprobat sistemul calității despre orice plan de modificare substanțială a sistemului calității sau a gamei de produse acoperite de acest sistem.
Organismul notificat trebuie să evalueze modificările propuse și să verifice dacă după operarea acestor modificări sistemul calității își menține conformitatea cu cerințele menționate la subpct. 2) din prezentul punct. Rezultatele evaluării argumentate și decizia privind rezultatul inspecției, care conține concluziile inspecției, urmează să fie comunicate producătorului.
4. Cerințele pentru examinarea proiectului produsului sînt următoarele:
1) pentru dispozitivele prevăzute în lista A din anexa nr. 2 la prezentul Regulament, suplimentar la obligațiile impuse la pct. 3 din prezenta anexă, producătorul depune la organismul notificat o cerere de examinare a dosarului de proiectare a dispozitivului pe care intenționează să îl fabrice și care intră în categoria menționată la pct. 3 subpct.1) din prezenta anexă;
2) cererea trebuie să descrie proiectul, procesul de fabricație și performanțele dispozitivului și să includă documentele indicate în pct. 3 subpct. 2) lit. c) din prezenta anexă pentru a aprecia dacă dispozitivul este în conformitate cu cerințele prezentului Regulament;
3) organismul notificat examinează cererea și dacă dispozitivul corespunde prevederilor prezentului Regulament, eliberează certificatul de examinare a proiectului. Organismul notificat poate solicita ca cererea să fie completată cu încercări sau cu probe suplimentare care să permită evaluarea conformității cu cerințele Regulamentului.
Certificatul va conține concluziile examinării, condițiile de validitate, datele necesare pentru identificarea proiectului aprobat și, după caz, descrierea scopului propus al dispozitivului;
4) modificările operate în proiectul aprobat trebuie să obțină în prealabil aprobarea organismului notificat care a emis certificatul de examinare a proiectului, dacă aceste modificări ar putea afecta conformitatea cu cerințele esențiale sau cu condițiile prevăzute pentru utilizarea dispozitivului. Solicitantul trebuie să informeze organismul notificat care a emis certificatul de examinare a proiectului despre orice schimbare efectuată în proiectul aprobat. Aprobarea adițională constituie un supliment la certificatul de examinare a proiectului;
5) producătorul este obligat să comunice fără întîrziere organismului notificat dacă a obținut informații despre orice schimbare a agenților patogeni sau a markerelor de infecție ce vor fi testați, în particular ca o consecință a variabilității și complexității biologice. În acest sens, producătorul va informa organismul notificat dacă o astfel de modificare ar putea afecta performanțele respectivului dispozitiv medical pentru diagnostic in vitro.
5. Supravegherea urmează să satisfacă următoarele cerințe:
1) scopul supravegherii este de a garanta că producătorul îndeplinește obligațiile impuse prin sistemul calității aprobat;
2) producătorul permite organismului notificat să efectueze inspecțiile necesare și îi furnizează orice informații relevante în legătură cu:
a) documentația privind sistemul calității;
b) datele prevăzute în sistemul calității cu privire la proiect, cum ar fi rezultatele analizelor, calculelor, încercărilor etc.;
c) datele prevăzute în sistemul calității cu privire la producție, cum ar fi rapoartele de inspecție, datele de testare și calibrare, rapoartele privind calificarea personalului;
3) organismul notificat efectuează periodic inspecții și evaluări pentru a se asigura că producătorul aplică sistemul calității aprobat și îi transmite producătorului un raport de inspecție;
4) suplimentar, organismul notificat face vizite inopinate producătorului, în cadrul cărora cere să se efectueze încercări pentru verificarea aplicării corecte a sistemului calității. Acesta furnizează producătorului un raport de inspecție sau de încercări, după caz.
6. Verificarea produselor fabricate prevăzute în lista A din anexa nr. 2 la prezentul Regulament se efectuează în felul următor:
1) în cazul dispozitivelor prevăzute în lista A din anexa nr. 2, producătorul trebuie să înainteze fără întîrziere organismului notificat, după încheierea controalelor și testelor, rapoartele asupra testelor realizate pe dispozitivele fabricate sau pe fiecare lot de dispozitive. Suplimentar, producătorul pune la dispoziția organismului notificat mostre ale dispozitivelor sau ale loturilor de dispozitive fabricate în conformitate cu condițiile și modalitățile prestabilite;
2) producătorul are dreptul să introducă dispozitivele pe piață dacă organismul notificat nu îi comunică în termenul prestabilit, dar nu mai tîrziu de expirarea a 30 de zile de la data primirii mostrelor, o altă decizie, incluzînd în particular orice condiție de validitate a certificatelor emise.
Anexa nr. 5
la Regulamentul privind condițiile de introducere
pe piață a dispozitivelor medicale
pentru diagnostic in vitro
la Regulamentul privind condițiile de introducere
pe piață a dispozitivelor medicale
pentru diagnostic in vitro
EXAMINAREA CE DE TIP
1. Examinarea CE de tip este procedura prin care un organism notificat constată și certifică faptul că o mostră reprezentativă din producția respectivă îndeplinește prevederile prezentului Regulament.2. Cererea pentru examinarea de tip va fi depusă de producător ori de reprezentantul său autorizat la un organism notificat și include:
1) numele și adresa producătorului și ale reprezentantului autorizat, dacă cererea este depusă de acesta din urmă;
2) documentația menționată la pct. 3 din prezenta anexă, necesară pentru evaluarea conformității mostrei reprezentative (în continuare – tip) cu cerințele prezentului Regulament. Solicitantul trebuie să prezinte un astfel de tip organismului notificat, iar acesta cere și alte mostre, după necesitate;
3) declarația scrisă prin care se menționează că examinarea aceluiași tip nu a fost solicitată altui organism notificat.
3. Documentația trebuie să permită înțelegerea proiectului, a procesului de fabricare și a performanțelor dispozitivului și să cuprindă în special următoarele aspecte:
1) descrierea generală a tipului, inclusiv toate variantele planificate;
2) toată documentația prevăzută la pct. 3 subpct. 3)-13) din anexa nr. 3 la prezentul Regulament;
3) în cazul dispozitivelor pentru autotestare, informațiile menționate la pct. 6 și 7 din anexa nr. 3 la prezentul Regulament.
4. Organismul notificat trebuie:
1) să examineze și să aprobe documentația, să verifice dacă tipul a fost fabricat în conformitate cu această documentație; să înregistreze produsele proiectate în conformitate cu prevederile aplicabile ale standardelor la care se referă pct. 18 din prezentul Regulament, precum și produsele care nu au fost proiectate conform acestor standarde;
2) să efectueze sau să organizeze inspecțiile și încercările necesare pentru a verifica soluțiile adoptate de producător și dacă respectă cerințele esențiale din prezentul Regulament, în cazul în care standardele la care se referă pct. 18 din prezentul Regulament nu au fost aplicate. Dacă dispozitivul este destinat să fie asociat cu alte dispozitive pentru a fi utilizat conform scopului propus, trebuie aduse dovezi că acesta corespunde cerințelor esențiale atunci cînd este asociat cu astfel de dispozitive avînd caracteristicile specificate de către producător;
3) să efectueze sau să solicite inspecțiile și încercările necesare pentru a verifica, în cazul în care producătorul a decis să aplice standardele relevante, dacă acestea au fost într-adevăr aplicate;
4) să stabilească, de comun acord cu solicitantul, locul unde vor fi efectuate inspecțiile și încercările necesare.
5. Dacă tipul este conform cu prevederile prezentului Regulament, organismul notificat emite un certificat de examinare CE de tip. Certificatul va conține numele și adresa producătorului, concluziile inspecției, condițiile de validitate și datele necesare pentru identificarea tipului aprobat. Documentația relevantă este anexată la certificat, iar o copie se păstrează la organismul notificat.
6. Producătorul este obligat să informeze fără întîrziere organismul notificat dacă a obținut informații despre orice modificări ale agenților patogeni și ale markerelor infecțiilor ce urmează a fi testați, în particular ca o consecință a variabilității și complexității biologice. În acest sens, producătorul va informa organismul notificat dacă o astfel de modificare ar putea afecta performanțele respectivului dispozitiv medical pentru diagnostic in vitro.
7. Modificările dispozitivului aprobat trebuie să obțină aprobarea suplimentară a organismului notificat care a emis certificatul de examinare are CE de tip, dacă aceste modificări afectează conformitatea cu cerințele esențiale sau cu condițiile de utilizare prescrise. Solicitantul va informa organismul notificat care a emis certificatul de examinare de tip despre orice modificare a dispozitivului aprobat. Noua aprobare se emite sub forma unui supliment la certificatul inițial de examinare CE de tip.
8. Alte organisme notificate pot obține o copie de pe certificatul de examinare CE de tip și/sau de pe suplimentele acestuia. Anexele la certificat trebuie să fie accesibile și altor organisme notificate, la solicitarea argumentată a acestora, după informarea prealabilă a producătorului.
Anexa nr. 6
la Regulamentul privind condițiile de introducere
pe piață a dispozitivelor medicale
pentru diagnostic in vitro
la Regulamentul privind condițiile de introducere
pe piață a dispozitivelor medicale
pentru diagnostic in vitro
VERIFICAREA CE
1. Verificarea CE este procedura prin care producătorul ori reprezentantul său autorizat asigură și declară că produsele care fac obiectul procedurii prevăzute la pct. 4 din prezenta anexă sînt conforme cu tipul descris în certificatul de examinare de tip și cu cerințele aplicabile lor din prezentul Regulament.2. Producătorul trebuie să ia toate măsurile necesare pentru a se asigura că din procesul de fabricație rezultă produse conforme cu tipul descris în certificatul de examinare de tip și cu cerințele prezentului Regulament. Înaintea începerii fabricației, producătorul trebuie să își întocmească documentele care definesc procesul de fabricație, în special privind sterilizarea și caracterul adecvat al materiilor prime, dacă este necesar, și să definească procedurile necesare de testare la nivelul tehnologiilor de vîrf. Toate prescripțiile de rutină prestabilite urmează să fie implementate pentru a se asigura omogenitatea producției și conformitatea produselor cu tipul descris în certificatul de examinare de tip și cu cerințele prezentului Regulament ce le sînt aplicabile.
În măsura în care pentru anumite aspecte testarea finală, conform pct. 6 subpct. 3) din prezenta anexă, nu este adecvată, testarea, monitorizarea și metodele de control adecvate ale proceselor de fabricație urmează să fie stabilite de către producător, cu aprobarea organismului notificat. Prevederile pct. 5 din anexa nr. 4 la prezentul Regulament se aplică corespunzător procedurilor sus-menționate.
3. Producătorul trebuie să instituie și să actualizeze permanent proceduri sistematice de valorificare a experienței obținute privind dispozitivele în faza de postproducție și să implementeze măsurile corespunzătoare pentru aplicarea oricărei acțiuni corective necesare și pentru notificare, după cum se menționează la pct. 5 din anexa nr. 3 la prezentul Regulament.
4. Organismul notificat trebuie să efectueze examinările și încercările necesare ținînd seama de pct. 2 din prezenta anexă, pentru verificarea conformității produsului cu cerințele prezentului Regulament, fie prin examinarea și încercarea fiecărui produs, după cum se menționează la pct. 5 din prezenta anexă, fie prin examinarea și încercarea statistică, conform pct. 6 din prezenta anexă, la decizia producătorului. La realizarea verificării statistice conform pct. 6, organismul notificat decide cînd trebuie aplicate procedurile statistice pentru inspectarea fiecărui lot în parte sau pentru inspectarea de loturi izolate. O astfel de decizie se ia prin consultarea cu producătorul.
Dacă efectuarea examinărilor și încercărilor pe baze statistice nu este adecvată, examinările și încercările se pot face în mod aleatoriu, cu condiția ca o astfel de procedură asociată cu măsurile adoptate în conformitate cu pct. 2 alineatul doi din prezenta anexă să asigure un nivel echivalent de conformitate.
5. Verificarea prin examinarea și încercarea fiecărui produs se efectuează respectîndu-se următoarele cerințe:
1) fiecare produs este examinat individual și se efectuează încercările necesare definite în standardele relevante menționate în pct. 18 din prezentul Regulament sau alte încercări echivalente pentru verificarea conformității produsului cu tipul descris în certificatul de examinare de tip și cu cerințele prezentului Regulament;
2) organismul notificat trebuie să aplice sau să asigure că se aplică numărul său de identificare pe fiecare produs aprobat și să emită în scris un certificat de conformitate privind încercările efectuate.
6. Cerințele față de verificările statistice sînt următoarele:
1) producătorul trebuie să prezinte produsele fabricate sub formă de loturi omogene;
2) se iau, la întîmplare, una sau mai multe mostre din fiecare lot. Produsele care alcătuiesc mostra sînt examinate conform standardelor relevante și se efectuează testările adecvate, definite în standardele menționate la pct. 18 din prezentul Regulament, sau încercările echivalente pentru verificarea conformității produselor cu tipul descris în certificatul de examinare de tip și cu cerințele prezentului Regulament, pentru a stabili dacă lotul se acceptă sau se respinge;
3) procedura de control statistic al produselor se bazează pe atribute și/sau variabile, necesitînd metode de prelevare a mostrelor cu caracteristici operaționale care să asigure un grad înalt de siguranță și performanță, ținînd cont de nivelul actual de dezvoltare a tehnologiei de vîrf în domeniu. Metoda de prelevare a mostrelor este stabilită prin standardele menționate în pct. 18 din prezentul Regulament, ținîndu-se cont de natura specifică a categoriilor de produse respective;
4) dacă lotul este acceptat, organismul notificat aplică sau se asigură că se aplică numărul său de identificare pe fiecare produs și eliberează un certificat de conformitate privind încercările efectuate. Toate produsele unui lot sînt introduse pe piață, cu excepția cazului în care mostrele au fost necorespunzătoare;
5) dacă un lot este respins, organismul notificat trebuie să ia măsurile necesare pentru a preveni introducerea pe piață a lotului respectiv. În eventualitatea respingerii frecvente a loturilor, organismul notificat suspendă verificarea statistică;
6) producătorul este în drept, sub responsabilitatea organismului notificat, să aplice numărul de identificare al acestuia în timpul procesului de fabricație.
Anexa nr. 7
la Regulamentul privind condițiile de introducere
pe piață a dispozitivelor medicale
pentru diagnostic in vitro
la Regulamentul privind condițiile de introducere
pe piață a dispozitivelor medicale
pentru diagnostic in vitro
DECLARAȚIA DE CONFORMITATE CE
Asigurarea calității producției
1. Producătorul trebuie să asigure aplicarea unui sistem al calității aprobat pentru fabricarea dispozitivelor, efectuarea inspecției finale, după cum se specifică la pct. 3 din prezenta anexă, fiind subiectul supravegherii prevăzute la pct. 5 din anexa nr. 4 la prezentul Regulament.Asigurarea calității producției
2. Declarația de conformitate este parte a procedurii prin care producătorul care îndeplinește obligațiile impuse la pct. 1 din prezenta anexă asigură și declară că produsele respective sînt conforme cu tipul descris în certificatul de examinare de tip și corespund dispozițiilor aplicabile ale prezentului Regulament.
Producătorul trebuie să aplice marcajul CE în conformitate cu prevederile prezentului Regulament și să întocmească o declarație scrisă de conformitate cu privire la dispozitivele respective.
3. Sistemul calității este organizat în felul următor:
1) producătorul trebuie să depună la un organism notificat o cerere pentru evaluarea sistemului calității, care va cuprinde:
a) toate documentele și mențiunile specificate la pct. 3 subpct.1) din anexa nr. 4 la prezentul Regulament;
b) documentația tehnică a tipurilor aprobate și copia de pe certificatul de examinare CE de tip;
2) aplicarea sistemului calității trebuie să asigure că dispozitivele sînt conforme cu tipul descris în certificatul de examinare CE de tip.
Toate elementele, cerințele și prevederile adoptate de producător pentru sistemul său de calitate trebuie să fie documentate în mod sistematic și ordonat sub formă de proceduri și declarații scrise privind politica de calitate. Documentația sistemului de calitate trebuie să permită interpretarea uniformă a politicii de calitate și a procedurilor, cum ar fi planuri, programe, manuale și înregistrări de calitate.
Documentația trebuie să cuprindă, în special, o descriere adecvată:
a) a obiectivelor producătorului privind sistemul calității;
b) a modului de organizare a activității și, în particular:
- structurile organizatorice, responsabilitățile echipei de conducere și a autorității organizatorice în ceea ce privește calitatea proiectării și fabricării dispozitivelor;
- metodele de monitorizare a eficienței sistemului calității și, în particular, capacitatea lui de a atinge calitatea dorită în proiectare și în producere, inclusiv controlul dispozitivelor ce nu se conformează cerințelor;
c) a tehnicilor de inspecție și de asigurare a calității în stadiul de producție, în particular:
- procesele și procedurile care vor fi utilizate, în special cu privire la sterilizare;
- procedurile legate de achiziționare;
- procedurile de identificare a produsului, schițate și actualizate prin proiecte, specificații sau alte elemente relevante, în orice stadiu de producție;
d) a încercărilor și verificărilor care trebuie să fie efectuate înainte, în timpul și după fabricație, a frecvenței cu care vor avea loc și a echipamentelor de testare utilizate; trebuie să fie asigurată trasabilitatea măsurărilor;
3) organismul notificat trebuie să verifice sistemul calității pentru a determina dacă aceasta satisface cerințele menționate la pct. 3 subpct. 2) din prezenta anexă. Se presupune că sistemul de calitate care implementează standardele relevante este conform cu aceste cerințe.
Echipa de evaluare trebuie să aibă, în mod obligatoriu, experiență în evaluarea tehnologiilor respective. Procedura de evaluare trebuie să includă o inspecție la locul de producție și, în cazuri justificate, la locul de producție al furnizorilor și/sau al subcontractanților, pentru a inspecta procesul de fabricație.
Decizia inspecției, care conține o evaluare argumentată și concluziile acestuia, este comunicată producătorului;
4) producătorul informează organismul notificat care a aprobat sistemul calității asupra oricărui plan de modificare substanțială a sistemului calității.
Organismul notificat trebuie să evalueze modificările propuse și să verifice dacă după operarea acestor modificări sistemul calității își menține conformitatea cu cerințele menționate la pct. 3 subpct. 2) din prezenta anexă. Decizia privind rezultatul inspecției care conține concluziile inspecției și o evaluare argumentată, este comunicată producătorului;
5) supravegherea se aplică conform prevederilor pct. 5 din anexa nr. 4 la prezentul Regulament.
4. Verificarea produselor prevăzute în lista A din anexa nr. 2 la prezentul Regulament se realizează după cum urmează:
1) producătorul înaintează, fără întîrziere, organismului notificat, ținînd cont de concluziile controalelor și testelor, rapoartele asupra testelor realizate pe dispozitivele fabricate sau pe fiecare lot de dispozitive. Suplimentar, producătorul pune la dispoziția organismului notificat mostre de dispozitive sau ale loturilor de dispozitive fabricate în conformitate cu condițiile și modalitățile prestabilite;
2) producătorul poate introduce dispozitivele pe piață dacă organismul notificat nu îi comunică în termenul prestabilit, dar nu mai tîrziu de expirarea a 30 de zile de la recepționarea mostrelor, o altă decizie, incluzînd în particular orice condiție de validitate a certificatelor emise.
Anexa nr. 8
la Regulamentul privind condițiile de introducere
pe piață a dispozitivelor medicale
pentru diagnostic in vitro
la Regulamentul privind condițiile de introducere
pe piață a dispozitivelor medicale
pentru diagnostic in vitro
DECLARAȚII ȘI PROCEDURI
privind dispozitivele destinate evaluării performanței
1. Pentru dispozitivele destinate evaluării performanței, producătorul ori reprezentantul său autorizat va întocmi o declarație conform prevederilor pct. 2 din prezenta anexă și va garanta că prevederile relevante ale prezentului Regulament sînt îndeplinite.privind dispozitivele destinate evaluării performanței
2. Declarația va conține următoarele informații:
1) datele ce permit identificarea dispozitivului respectiv;
2) planul de evaluare prin care se stabilesc scopul, bazele științifice, tehnice sau medicale, domeniul evaluării și numărul de dispozitive vizate;
3) lista laboratoarelor sau altor instituții care participă la studiul de evaluare;
4) data de pornire și durata programului de evaluare, iar în cazul dispozitivelor pentru autotestare, locul și numărul persoanelor neprofesioniste implicate;
5) declarația din care să rezulte că dispozitivul în cauză este conform cerințelor prezentului Regulament, cu excepția aspectelor acoperite de evaluare și a celor special menționate în declarație, și că au fost luate toate măsurile pentru protecția sănătății și siguranței pacientului, a utilizatorului și a altor persoane.
3. Producătorul trebuie să păstreze la dispoziția Agenției documentația care să permită înțelegerea proiectului, a procesului de fabricație și a performanțelor produsului, inclusiv a performanțelor preconizate, astfel încît să permită evaluarea conformității cu cerințele impuse de prezentul Regulament. Această documentație urmează să fie păstrată pentru o perioadă de cel puțin 5 ani de la data încheierii evaluării performanței.
Producătorul va lua toate măsurile necesare pentru asigurarea procesului de fabricație, astfel încît produsele să fie conforme cu documentația menționată în primul punct.
4. Prevederile prezentului Regulament se aplică și dispozitivelor destinate evaluării performanței.
Anexa nr. 9
la Regulamentul privind condițiile de introducere
pe piață a dispozitivelor medicale
pentru diagnostic in vitro
la Regulamentul privind condițiile de introducere
pe piață a dispozitivelor medicale
pentru diagnostic in vitro
CRITERII PENTRU DESEMNAREA
ORGANISMELOR DE EVALUARE A CONFORMITĂȚII NOTIFICATE
1. Proiectantul, producătorul, furnizorul, instalatorul sau utilizatorul dispozitivelor pe care le inspectează, precum și reprezentanții autorizați ai vreuneia dintre aceste persoane nu au dreptul să funcționeze în calitate de organism de evaluare a conformității notificat, conducător al acestuia și membru al personalului de evaluare și verificare.ORGANISMELOR DE EVALUARE A CONFORMITĂȚII NOTIFICATE
Aceștia nu pot să fie direct implicați în proiectarea, construcția, introducerea pe piață sau întreținerea dispozitivelor și nici să reprezinte părțile angajate în astfel de activități. Aceasta însă nu exclude în niciun mod posibilitatea schimbului de informații tehnice între producător și organismul de evaluare a conformității notificat.
2. Organismul notificat și personalul său efectuează evaluarea și verificarea la cel mai înalt nivel de integritate profesională și competență în domeniul dispozitivelor medicale și este liber de orice presiune și influență, în special financiară, care le-ar putea influența decizia privind rezultatele inspecției, în special din partea persoanelor sau a grupurilor de persoane interesate de rezultatul verificărilor.
Dacă organismul de evaluare a conformității notificat subcontractează sarcini specifice în legătură cu stabilirea și verificarea faptelor, acesta se asigură mai întîi că subcontractantul respectă cerințele prezentului Regulament și, în special, pe cele ale prezentei anexe. Organismul de evaluare a conformității notificat păstrează la dispoziția Ministerul Sănătății, Muncii și Protecției Sociale documentele relevante de evaluare a calificărilor subcontractantului și cele privind activitatea acestuia care cad sub incidența prezentului Regulament.
3. Organismul de evaluare a conformității notificat este capabil să execute toate sarcinile atribuite acestor tipuri de organisme prin una dintre anexele nr. 3-7 la prezentul Regulament, sarcini pentru care a fost notificat, indiferent dacă aceste sarcini sînt îndeplinite de însuși organismul respectiv sau doar pe răspunderea lui. În special, organismul notificat dispune de personalul și de facilitățile necesare pentru îndeplinirea în mod corespunzător a sarcinilor tehnice și administrative aferente evaluării și verificării.
Acest lucru presupune existența în cadrul organizației a unui număr suficient de personal științific, care să posede experiență și cunoștințe suficiente pentru a evalua funcționalitatea medicală și performanța dispozitivelor pentru care a fost notificat, avînd în vedere cerințele prezentului Regulament, în special cele enunțate în anexa nr. 1 la Regulament.
Organismul de evaluare a conformității notificat trebuie, de asemenea, să aibă acces la echipamentul necesar pentru verificările cerute.
4. Personalul organismului de evaluare a conformității notificat are:
1) instruire profesională temeinică pentru operațiunile de evaluare și verificare pentru care organismul a fost notificat;
2) cunoștințe suficiente în domeniul reglementărilor cu privire la inspecțiile pe care le efectuează și o experiență adecvată;
3) capacitate adecvată pentru întocmirea certificatelor, a înregistrărilor și a rapoartelor ce demonstrează efectuarea inspecțiilor.
5. Imparțialitatea organismului de evaluare a conformității notificat este garantată. Salarizarea personalului acestuia nu depinde de numărul inspecțiilor efectuate și nici de rezultatele inspecțiilor.
6. Organismul de evaluare a conformității notificat are o asigurare de răspundere civilă, cu excepția cazului în care autoritatea competentă însăși efectuează inspecțiile în mod direct.
7. Personalul organismului de evaluare a conformității notificat este obligat să respecte secretul profesional cu privire la toate informațiile obținute în exercitarea atribuțiilor sale (cu excepția raporturilor cu autoritățile publice competente ale statului) conform prezentului Regulament sau oricărei prevederi legale în vigoare ce reglementează domeniul dispozitivelor medicale.
Anexa nr. 10
la Regulamentul privind condițiile de introducere pe piață a dispozitivelor medicale
pentru diagnostic in vitro
la Regulamentul privind condițiile de introducere pe piață a dispozitivelor medicale
pentru diagnostic in vitro
MARCAJUL CE
1. Marcajul CE constă în inițialele „CE” cu următoarea formă: 3. Diferitele componente ale marcajului CE au în principal aceeași dimensiune verticală, care nu poate fi mai mică de 5 mm.
4. La această dimensiune minimă se poate renunța în cazul dispozitivelor fabricate în serie mică.